Abstract
Sulfuretin is a natural flavonoid found in the plant Rhus verniciflua STOKES. The plant has been traditionally used as medicinal agent for antiviral, cathartic, diaphoretic, anti-rheumatic and sedative activities in East Asia. In this study we isolated and identified sulfuretin from R. verniciflua and investigated its anti-adipogenic activity against 3T3-L1 preadipocytes cells. We evaluated the effects of sulfuretin on the adipogenic transcription factors like peroxisome proliferator-activated receptor γ (PPARγ), CCAAT/enhancer-binding protein α (C/EBPα), fatty acid synthase (FAS), Fabp4, adiponectin and zinc fingerprint protein (Zfp) 521 by gene expression (real-time QPCR) and Western blot analysis. Sulfuretin treatment at Day 0 and 2 showed significant reduction of lipid production in 3T3-L1 cells in concentration dependent manner. Gene expression analysis (real-time PCR) revealed that sulfuretin inhibited the both major adipogenic factors (C/EBPα, C/EBPβ and PPARγ) and minor adipogenic factors (sterol regulatory element-binding protein (SREBP1c), adiponectin, FAS, Fabp4, Zfp423, and Ebf1). Western blot analysis showed the increased expression of β-catenin and suppression of PPARγ after sulfuretin treatment. Overall, sulfuretin is a natural flavonoid having potent anti-adipogenic activity through the suppression of major adipogenic factors C/EBPα, C/EBPβ and PPARγ, which initiate adipogenesis.
Obesity is emerging as a burning health problem around the world. Despite various treatment measures for obesity, a low fat diet and increased physical activity is the main treatment approach.1) However, Pharmacotherapy is necessary or recommended for those having a body mass index (BMI) ≥30 kg/m2 or with a BMI ≥27 kg/m2 along with the presence of two or more obesity-related complications like coronary heart disease, type-2 diabetes, or sleep apnea.2)
Obesity, a major risk factor for cardiovascular disease, high blood pressure, type-2 diabetes and carcinogenesis, is mainly characterized by the increased adipogenesis, which is contributed by increase in both the number and size of the adipose cells.3–6) Adipogenesis, which involves various cellular processes, gene expressions and hormones, brings changes in cellular morphology of pre-adipocytes and converts them to adipocytes which can store large amount of fat.7,8) Many of the transcription factors, such as peroxisome proliferator-activated receptor γ (PPARγ), the CCA AT/enhancer-binding protein (C/EBP) family, sterol regulatory element-binding protein (SREBP1c), adiponectin, fatty acid synthase (FAS), zinc fingerprint protein (Zfp423), and early B cell factor 1 (Ebf1) significantly play roles in adipogenesis either directly or indirectly. PPARγ, C/EBP, and SREBP-1c are important regulators at the initial stage of adipogenesis.9)
Rhus verniciflua STOKES (family: Anacardiaceae) has been traditionally used as medicine for antiviral, cathartic, diaphoretic, anti-rheumatic and sedative activities in East Asia.10) In a study, the R. verniciflua extract has significantly reduced body weight, total cholesterol and low density lipoprotein (LDL)-cholesterol level in high-fat diet mice.10) In this study, we isolated a flavonoid compound, sulfuretin from the plant R. verniciflua and studied its anti-adipogenic activity on 3T3-L1 cells. Different studies have already reported its biological activities: anti-oxidative,11) anti-rheumatic,12) antimutagenic,13) cell protective,14) and anti-cancer activity.15) Its anti-adipogenic study is reported for the first time in this study.
EXPERIMENTAL
MaterialsAerial parts of R. verniciflua were bought form Human Herb Co., Ltd., S. Korea. The voucher specimens (10-022RV) were deposited in the Pharmacognosy Laboratory of Wonkwang University, South Korea. Silica-gel (Kieselgel 60, 70–230 and 230–400 mesh, Merck, Darmstadt, Germany) and ODS-A (12 nm, S-75 µm, YMC, Kyoto, Japan) were used for column chromatography. Solvents including chloroform (CHCl3), ethanol (EtOH), methanol (MeOH), acetone, ethyl acetate (EtOAc), hexane, n-butanol, were purchased form SK chemicals (Seongnam, Korea) and were of HPLC grade. 3T3-L1 cells were obtained from the American Type Culture Collection (Rockville, MD, U.S.A.). 1H-, 13C-NMR and two-dimensional NMR (2D-NMR) were recorded on JEOL Eclipse 500 FT-NMR spectrometer (1H-NMR, 500 MHz; 13C-NMR, 125 MHz) using deuterated solvent (dimethyl sulfoxide (DMSO)-d6 containing 0.03% tetramethylsilane (TMS)). High resolution electrospray ionization (ESI) mass spectra were obtained on an Agilent 6120 mass spectrometer. Dexamethasone, 1-methy-3-isobutylxanthine (IBMX), Dulbecco’s modified Eagle’s medium (DMEM), insulin were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Antibodies against PPARγ, β-catenin, secondary antibodies were from Santa Cruz Bio-technology. Trizol reagent, RT-PCR kit and primers were bought from Invitrogen (Carlsbad, CA, U.S.A.).
Isolation and IdentificationR. verniciflua leaves were dried and extracted with methanol by heating (40°C). The solvent was removed using rotatory evaporator and the dry methanol extract was suspended in water and fractionated with butanol. Dried butanol fraction was loaded on silica gel open column and eluted with gradient (CHCl3)–MeOH (100→50%) to obtain fractions F1 to F10. Fraction 6 was again subjected to column chromatography on reverse phase silica gel (Octadecylsilane (ODS) chromatography) using solvent system of MeOH and water (50 to 70% MeOH). A pure compound (orange amorphous powder) was isolated which was later identified as sulfuretin after the UV spectrum, 1H- and 13C-NMR study.
Ultra Performance Liquid Chromatography (UPLC) AnalysisThe stock solution was prepared in methanol at a concentration of 5 mg/mL for methanol extract of R. verniciflua and 1 mg/mL for sulfuretin. All the solutions prior to injection in UPLC machine were filtered through 0.20 µm filters (polytetrafluoroethylene (PTFE), hydrophilic filter). UPLC analysis was conducted on an Agilent 1290 series LC system (Agilent Technologies, CA, U.S.A.), which consisted DEBAA03145 binary pump, DEBAF02229 diode array detector, a DEBAP03582 auto sampler. The chromatographic signals were analyzed by Agilent ChemStation software version 1.3. Halo RP-amide column (2.7, 4.6 150 mm) with solvent system methanol (A) and 0.1% phosphoric acid (B) was used for chromatographic separation. The gradient solvent system was optimized as: 0–20 min, 0–100% A at a flow rate of 1 mL/min. The column temperature was maintained at 30°C and injection volume was 5 µL. The detection was conducted at 210 nm. For the calibration curve, a serial dilution of the stock solution of sulfuretin (1000–15.625 µg/mL) was done to prepare the calibration curve. Five concentrations of sulfuretin were analysed in triplicate for the establishment of calibration curves (plotting the concentrations of analyte vs. respective peak areas). The equation of calibration curve was used for the quantification of sulfuretin in the plant R. verniciflua.
Cell Viability AssayThe 3T3-L1 preadipocytes were cultured in 96 well plates and incubated until 100% confluency. The cells were then treated with various concentration of sulfuretin (20–150 µM) for 48 h followed by the treatment with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution for three hours. The formazan complex was dissolved with DMSO and the absorbance was measured in enzyme-linked immunosorbent assay (ELISA) reader.
Culture and Differentiation of 3T3-L1 Cells3T3-L1 pre-adipocytes were cultured in DMEM with 10% bovine calf serum at 37°C in a humidified atmosphere of 5% CO2. Two days after the 100% confluency (Day 0), cell were stimulated to differentiation by replacing with differentiation media (MDI), i.e., DMEM containing 10% fetal bovine serum (FBS), 0.5 mM IBMX, 1 µM dexamethasone and 10 µg/mL insulin. After two days (Day 2), the culture media was replaced with DMEM containing 10% FBS and 10 µg/mL insulin. After that the media was changed every 2 d (Day 4, 6 and 8) with DMEM containing only 10% FBS. The cells were treated with non-toxic concentration of sulfuretin (100, 70 and 40 µM) at Day 0 and 2 to evaluate the anti-adipogenic activity. After the establishment of anti-adipogenic activity of sulfuretin, a single concentration (70 µM) was treated on Day −2 (two days before addition of differentiation media), Day 0, 2, 4, 6 and 8 to evaluate at which stage the treatment would show maximum inhibition of lipid production.
Oil Red O (ORO) StainingAt Day 10 ORO staining was done to determine the lipid production. Cells were washed twice with 1×phosphate buffered saline (PBS), fixed in 10% formaldehyde for 30 min, and then washed with 60% isopropanol and then stained with the Oil Red O working solution (6 : 4, 0.6% ORO dye in isopropanol–water) for 30 min at 25°C and washed three times with water. Staining was visualized by bright-field microscopy, and ORO dyes extracted from the cells in isopropanol were quantified at a wavelength of 520 nm.
Protein Extraction and Western Blotting3T3-L1 cells were differentiated with or without using sulfuretin (70 µM). Cells were collected at the indicated days (Day 0, 1, 3, 6 and 8) using a cell scraper. For the cell lysis, cells were treated with ice-cold RIPA buffer containing 25 mM Tris–HCl (pH 7.6), 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS) and a protease inhibitor cocktail (Sigma-Aldrich) for 30 min. All cell lysates were centrifuged at speed of 14000 rpm for 20 min at 4°C to remove cell debris. Then the protein concentration of each sample was calculated using a BCA protein assay kit (Pierce, Rockford, IL, U.S.A.). Cellular proteins (50 µg) were loaded and separated by 10% SDS-polyacrylamide gel electrophoresis. After that the gels were transferred to nitrocellulose membranes at 150 mA for 1 h and were blocked with PBS containing 5% skim milk and 0.1% Tween 20 for 2 h at room temperature. The blots were incubated with primary antibodies (1 : 1000 dilutions) overnight at 4°C followed by secondary antibody for 1 h. The protein bands were determined from the gel image system.
Quantitative Real-Time PCR3T3-L1 cells were differentiated in presence or absence of sulfuretin (70 µM). Cells were collected at the indicated days (Day 0, 1, 3, 6, 8 and 10) and lysed with 1 mL Trizol. The acquired cell lysate were added to chloroform and centrifuged to yield RNA fraction. The RNA fraction was mixed with 0.5 mL isopropyl alcohol and centrifuged at 12000 rpm for 10 min at 4°C for RNA precipitation. The precipitated RNA were washed and dried at room temperature for 10 min and dissolved in ribonuclease (RNase)-free diethyl pyrocarbonate (DEPC) treated water. The first strand cDNA was synthesized using a cDNA synthesis kit (Clontech Advantage® RT-for-PCR kit, #639506). The gene expression levels were analysed by quantitative real-time PCR using AB 7900HT Real Time PCR system (Applied Biosystems #4364346, Foster City, CA, U.S.A.). The primers used in the experiments are shown in Table 1. After an initial incubation for 2 min at 50°C, the cDNA was denatured at 95°C for 5 min followed by 40 cycles of PCR (95°C, 20 s, 60°C, 120 s). All results were obtained from at least three independent experiments. The mRNA levels of all genes were normalized using cyclophilin as internal control.
Table 1. The Primer Sequence Used for Real-Time PCR
| Gene name | Forward primer | Reverse primer |
|---|
| PPARγ | GCGGCTGAGAAATCACGTT | TCAGTGGTTCACCGCTTCTT |
| C/EBPα | GACCATTAGCCTTGTGTGTACTGTATG | TGGATCGATTGTGCTTCAAGTT |
| C/EBPβ | CGGGGTTGTTGATGTTTTTGG | CCGAAACGGAAAAGGTTCTCA |
| C/EBPδ | ACGACGAGAGCGCCATC | TCGCCGTCGCCCCAGTC |
| Adiponectin | GATCACTGTCAGCAGGACTT | TGCCTCTTCAAGTAGCTCA |
| FAS | GGT GAC ACT CGC AGA AGA CAA TA | AACAGCCTCAGAGCGACAAT |
| SREBP-1c | GCATGCCATGGGCAAGTAC | TGTTGCCATGGAGATAGCATCT |
| Fabp4 | CTTCAAACTGGGCGTGGAA | CTAGGGTTATGATGCTCTTCACCTT |
| Zfp423 | GATCACTGTCAGCAGGACTT | TGCCTCTTCAAGTAGCTCA |
| Ebf1 | TGCTGGTCTGGAGTGAGTTGA | CCACCACACCAGGGATGTG |
RESULTS AND DISCUSSION
Structure Identification of Isolated CompoundThe 1H-NMR, 13C-NMR, mass and UV-visible absorption maximum (λmax) data of the isolated compound were taken for the structure elucidation. The data as given below were compared with the published literatures and the compound was confirmed to be sulfuretin11,16,17) (Fig. 1).
Orange coloured powder, C15H10O5, MW 270. Electrospray ionization (ESI)-MS m/z (negative) 269.10 [M−H]−; 1H-NMR (500 MHz, DMSO-d6) δ: 6.64 (1H, s, H-2), 6.70 (1H, dd, J=1.85, 8.25 Hz, H-6), 6.74 (1H, d, J=1.85 Hz, H-8), 6.83 (1H, d, J=8.25 Hz, H-5′), 7.24 (1H, dd, J=2.3, 8.2 Hz, H-6′), 7.44 (1H, d, J=1.85 Hz, H-2′), 7.60 (1H, d, J=8.7 Hz, H-5); 13C-NMR (125 MHz, DMSO-d6) δ: 112.3 (C-2), 146.2 (C-3), 148.6 (C-4), 126.3 (C-5), 125.1 (C-6), 168.0 (C-7), 98.9 (C-8), 166.7 (C-9), 113.8 (C-10), 123.9 (C-1′), 118.5 (C-2′), 146.1 (C-3′), 148.6 (C-4′), 116.6 (C-5′), 125.1 (C-6′). UV/V λmax (MeOH): 206, 210, 394 nm.
UPLC AnalysisThe UPLC analysis was used for the quantification of the sulfuretin in the plant R. verniciflua. The retention time of isolated compound and its UV spectrum was evaluated for the identification of sulfuretin peak in the chromatogram of methanol extract of R. verniciflua. The UPLC chromatograms of sulfuretin and plant extract (Fig. 2) reveal sulfuretin peak at retention time of approximately 10.35 min. The chromatograms were detected at 210 nm wavelength. The linearity of the calibration curve was satisfactory (R2=0.996). The quantification of sulfuretin in the R. verniciflua was obtained from the regression equation of the calibration curve and peak area of sulfuretin in the extract. The R. verniciflua contains around 0.122% of sulfuretin (Table 2).
Table 2. Quantification Table of Sulfuretin in
R. verniciflua from the Calibration Curve
| Analyte | Regression equation | Correlation coefficient | Quantity (mg/g of plant) |
|---|
| Sulfuretin | y=18009x+280.02 | 0.9966 | 1.22 |
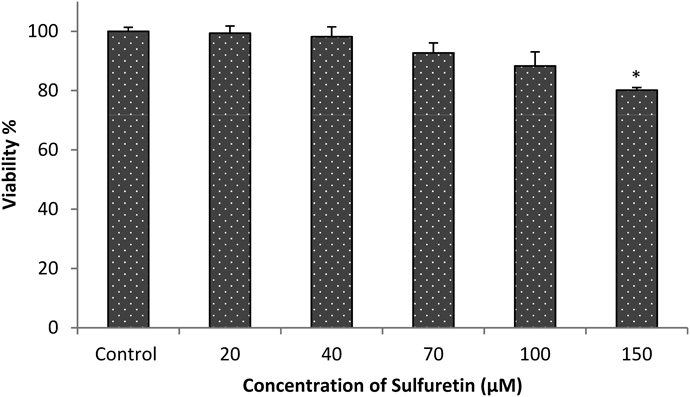
Different concentrations of sulfuretin (20–150 µM) were taken for the viability test against 3T3-L1 cells using MTT assay. The results of viability test (Fig. 3), show that up to 100 µM of sulfuretin was safe (nearly 90% viability) for the 3T3-cell experiment.
Effect of Sulfuretin in the Accumulation of Lipid Droplets in 3T3-L13T3-L1 preadipocytes cell were differentiated with or without the presence of sulfuretin (100, 70 and 40 µM) as indicated in Fig. 4A. At Day 10 the lipid production was evaluated and quantified by ORO staining technique. The lipid droplets (red) as shown in Fig. 4B captured in microscope reveal that sulfuretin strongly inhibited the lipid production. The lipid production was reduced by approximately 65% at 70 and 100 µM (Fig. 4C). It confirmed that sulfuretin inhibits lipid production in 3T3-L1 cells significantly when treated at Day 0 and 2.
We also tried to find out the anti-adipogenic effect of sulfuretin, when treated before the beginning of differentiation (Day −2) and at the later part of differentiation (Day 4, 6 and 8) as indicated in Fig. 5A. The ORO results (Fig. 5B) revealed that sulfuretin treatment at Day 0 and 2 only had significant inhibition on adipogenesis. There was no effect of sulfuretin when the treatment was at Day −2, i.e., two days before the treatment of differentiation media. Similarly sulfuretin treatment after some days of introducing differentiation media (Day 4, 6 and 8) had also no good results in the inhibition of lipid production. Sulfuretin was found to be most effective when the treatment was done at Day 0 and 2. From these observations we can understand that sulfuretin inhibits the master adipogenic factors which initiate the adipogenesis. Once the adipogenesis is started, then the role of sulfuretin in lipid inhibition is rapidly diminished.
Effect of Sulfuretin on mRNA Levels of PPARγ, CCAAT/Enhancer Binding Protein (C/EBP), and β-CateninFrom several studies it has been established that C/EBPβ and C/EBPδ are the first transcription factors induced after exposure of MDI to the 3T3-L1 preadipocytes.18) Different study has provided the correlation of induction of PPARγ and C/EBPα during adipogenesis by C/EBPβ and C/EBPδ activity.19,20) The activity of C/EBPβ and C/EBPδ is thought to mediate the expression of PPARγ, which is transcriptionally induced two days after induction of differentiation and remain maximum from Day 6 to 8.21,22) In our study also (Figs. 6A–E) we saw similar pattern in the level of all the three adipogenic factors in the control during the adipogenesis. When sulfuretin was treated along with MDI (Day 0), we saw decrease in expression of C/EBPβ and C/EBPδ, the early transcription factors of adipogenesis. The expression of PPARγ and C/EBPα also decreased after sulfuretin treatment. These results suggest that sulfuretin blocked the expression of C/EBPβ and C/EBPδ which in turn inhibited the expression of PPARγ and C/EBPα. This illustrates that sulfuretin suppresses the early program of adipogenesis by inhibiting C/EBPβ and C/EBPδ. The ORO results also support this finding as there was greater inhibition of lipid production in early treatment of sulfuretin (Fig. 5B).
It has been well established that Wnt signalling is a negative regulator of adipogenesis in vitro and in vivo by interfering the induction of PPARγ and C/EBPα.23) Thus, we hypothesized that sulfuretin may inhibit the adipogenesis, also through the activation of Wnt pathways. To test that, we measured β-catenin protein levels as the readout for the activity of canonical Wnt signalling. From the Western blot data (Fig. 6E) we found that β-catenin levels were significantly increased with sulfuretin treatment. So, the activation of β-catenin by sulfuretin also medicated the suppression of PPARγ.
Effect of Sulfuretin on Expression of SREBP-1c, Adiponectin, FAS, Fabp4, Zfp423, and Ebf1Our next investigation was to see how sulfuretin brings changes in the level of other regulators of adipogenesis. SREBP-1c, adiponectin, FAS, Fabp4, Zfp423 and Ebf1 are the important regulators of production, transport and storage of lipid. All these factors have important role in lipid production and accumulation in 3T3-L1 cells. The role of SREBP-1c is to stimulate transcription of genes which involve in fatty acid synthesis.24) The enzyme FAS is a key enzyme of lipogenesis which catalyses all the reactions that produce palmitic acid from acetyl-CoA and malonyl-CoA.25) During the time of lipogenesis, its activity gets triggered.26) Adiponectin is another important protein for cell proliferation and differentiation from preadipocytes to adipocytes and play vital role in increasing lipid content in adipocytes.27) FABP4 is known to regulate intracellular fatty acid and lipid metabolisms.28) Ebf1 induces the expression of PPARγ and C/EBPα and Zfp423 promotes adipose linage commitment.29,30) We checked the level of expression of mRNAs of SREBP-1c, adiponectin, FAS, Fabp4, Zfp423 and Ebf1 using QPCR in the differentiated adipocytes with or without the treatment of sulfuretin at Day 0. The regulators were found to be highly expressed during adipogenesis (control group) and when sulfuretin was treated along with MDI, levels of mRNA expression of those regulators were decreased (Fig. 7). So, our data suggest that sulfuretin suppressed the expression of other regulators involved in lipid transportation and storage.
CONCLUSION
It is well accepted that the excessive synthesis and accumulation of lipid in the adipocyte cells leads to the occurrence of obesity.31) To treat obesity, the exploration of natural products as alternative medicine is increasing.32) This study is an endeavour to find and establish anti-adipogenic activity of sulfuretin. It inhibited the 3T3-L1 proliferation in a dose and time dependent manner by inhibiting early adipogenic factors like C/EBPβ and C/EBPδ along with β-catenin. Sulfuretin is a major constituent of the plant, R. verniciflua, which anti-adipogenic activity has already been reported. So, we can estimate that the anti-adipogenic activity of R. verniciflua might be due to sulfuretin. Overall, sulfuretin is a potent anti-adipogenic phytochemical and can be a potential pharmaceutical ingredient for obesity treatment. So, a further study on this flavonoid is necessary.
Acknowledgment
This research was supported through Basic Science Research Program by the National Research Foundation of Korea (NRF-2010-0024284), under Science and Technology, Ministry of Education.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Caterson ID. Obesity and its management. Australian Prescriber, 22, 12–16 (1999).
- 2) National Institutes of Health/National Heart Lung and Blood Institute. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: the evidence report. Obes. Res., 6, 51S–210S (1998).
- 3) Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA, 282, 1523–1529 (1999).
- 4) Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell, 104, 531–543 (2001).
- 5) Pi-Sunyer FX. The obesity epidemic: pathophysiology and consequences of obesity. Obes. Res., 10 (Suppl. 2), 97S–104S (2002).
- 6) Alessi MC, Lijnen HR, Bastelica D, Juhan-Vague I. Adipose tissue and atherothrombosis. Pathophysiol. Haemost. Thromb., 33, 290–297 (2005).
- 7) Farmer SR. Transcriptional control of adipocyte formation. Cell Metab., 4, 263–273 (2006).
- 8) Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol., 7, 885–896 (2006).
- 9) Naowaboot J, Chung CH, Pannangpetch P, Choi R, Kim BH, Lee MY, Kukongviriyapan U. Mulberry leaf extract increases adiponectin in murine 3T3-L1 adipocytes. Nutr. Res., 32, 39–44 (2012).
- 10) Jeong S-J, Park J-G, Kim S, Kweon HY, Seo S, Na D-S, Lee D, Hong CY, Na C-S, Dong M-S, Oh GT. Extract of Rhus verniciflua stokes protects the diet-induced hyperlipidemia in mice. Arch. Pharm. Res., 38, 2049–2058 (2015).
- 11) Lee J-C, Lim K-T, Jang Y-S. Identification of Rhus verniciflua Stokes compounds that exhibit free radical scavenging and anti-apoptotic properties. Biochimica et Biophysica Acta (BBA)–General Subjects, 1570, 181–191 (2002).
- 12) Young-Rae Lee J-KH. Hyoung-Won Koh, Kyu Yun Jang, Ju Hong Lee, Jin-Woo Park and Byung-Hyun Park: Sulfuretin, a major flavonid isolated from Rhus veniciflula, ameliorates experimental arthritis in mice. Life Sci., 90, 19–20 (2012).
- 13) Park K-Y, Jung G-O, Lee K-T, Choi J, Choi M-Y, Kim G-T, Jung H-J, Park H-J. Antimutagenic activity of flavonoids from the heartwood of Rhus verniciflua. J. Ethnopharmacol., 90, 73–79 (2004).
- 14) Song M-Y, Jeong G-S, Kwon K-B, Ka S-O, Jang H-Y, Park J-W, Kim Y-C, Park B-H. Sulfuretin protects against cytokine-induced β-cell damage and prevents streptozotocin-induced diabetes. Exp. Mol. Med., 42, 628–638 (2010).
- 15) Poudel S, Song J, Jin E-J, Song K. Sulfuretin-induced miR-30C selectively downregulates cyclin D1 and D2 and triggers cell death in human cancer cell lines. Biochem. Biophys. Res. Commun., 431, 572–578 (2013).
- 16) Jung MJ, Chung HY, Kang SS, Choi JH, Bae KS, Choi JS. Antioxidant activity from the stem bark of Albizzia julibrissin. Arch. Pharm. Res., 26, 458–462 (2003).
- 17) Zhang X, Boytner R, Cabrera JL, Laursen R. Identification of yellow dye types in pre-Columbian Andean textiles. Anal. Chem., 79, 1575–1582 (2007).
- 18) Ntambi JM, Young-Cheul K. Adipocyte differentiation and gene expression. J. Nutr., 130, 3122S–3126S (2000).
- 19) Morrison RF, Farmer SR. Hormonal signaling and transcriptional control of adipocyte differentiation. J. Nutr., 130, 3116S–3121S (2000).
- 20) Farmer S. Regulation of PPARγ activity during adipogenesis. Int. J. Obes., 29 (Suppl. 1), S13–S16 (2005).
- 21) Clarke SL, Robinson CE, Gimble JM. CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor γ2 promoter. Biochem. Biophys. Res. Commun., 240, 99–103 (1997).
- 22) Wu Z, Xie Y, Bucher N, Farmer SR. Conditional ectopic expression of C/EBP beta in NIH-3T3 cells induces PPAR gamma and stimulates adipogenesis. Genes Dev., 9, 2350–2363 (1995).
- 23) Hasani-Ranjbar S, Larijani B, Abdollahi M, S. H-R. B. L, M. A: A systematic review of the potential herbal sources of future drugs effective in oxidant-related diseases. Inflamm. Allergy Drug Targets, 8, 2–10 (2009).
- 24) Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest., 109, 1125–1131 (2002).
- 25) Hillgartner FB, Salati LM, Goodridge AG. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiol. Rev., 75, 47–76 (1995).
- 26) Boizard M, Le Liepvre X, Lemarchand P, Foufelle F, Ferré P, Dugail I. Obesity-related overexpression of fatty-acid synthase gene in adipose tissue involves sterol regulatory element-binding protein transcription factors. J. Biol. Chem., 273, 29164–29171 (1998).
- 27) Fu Y, Luo N, Klein RL, Garvey WT. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J. Lipid Res., 46, 1369–1379 (2005).
- 28) Gerbens F, Jansen A, van Erp AJ, Harders F, Meuwissen TH, Rettenberger G, Veerkamp JH, te Pas MF. The adipocyte fatty acid-binding protein locus: characterization and association with intramuscular fat content in pigs. Mamm. Genome, 9, 1022–1026 (1998).
- 29) Jimenez MA, Åkerblad P, Sigvardsson M, Rosen ED. Critical role for Ebf1 and Ebf2 in the adipogenic transcriptional cascade. Mol. Cell. Biol., 27, 743–757 (2007).
- 30) Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Transcriptional control of preadipocyte determination by Zfp423. Nature, 464, 619–623 (2010).
- 31) Akiyama T, Tachibana I, Shirohara H, Watanabe N, Otsuki M. High-fat hypercaloric diet induces obesity, glucose intolerance and hyperlipidemia in normal adult male Wistar rat. Diabetes Res. Clin. Pract., 31, 27–35 (1996).
- 32) Moro CO, Basile G, G. MCaB. Obesity and medicinal plants. Fitoterapia, 71 (Suppl. 1), S73–S82 (2000).