Abstract
Secretory and membrane proteins are synthesized in ribosomes, then mature in the endoplasmic reticulum (ER), but if ER function is impaired, immature defective proteins accumulate in the ER. This situation is called ER stress: in response, a defensive mechanism called the unfolded protein response (UPR) is activated in cells to reduce the defective proteins. During the UPR, the ER transmembrane sensor molecules inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and RNA-dependent protein kinase (PKR)-like ER kinase (PERK) are activated, stress signals are transduced to the outside of the ER, and various cell responses, including gene induction, occur. In ER-associated degradation (ERAD), one type of UPR, defective proteins are eventually expelled from the ER and degraded in the cytoplasm through the ubiquitin proteasome system. Since ER stress has been reported to have relationships with neurodegenerative diseases, diabetes, metabolic syndromes, and cancer, it is the focus of increased attention from the perspectives of elucidating pathogenic mechanisms, and in the development of therapeutics.
1. INTRODUCTION
Secretory and membrane proteins, such as hormones and transporters, respectively, account for as many as one-third of synthesized proteins. After these have been synthesized in the ribosomes, they are incorporated into the endoplasmic reticulum (ER) and then mature by receiving folding and various other modifications: proteolytic processing, N-linked glycosylation, disulfide bond formation, etc. Only the completed items are sent to the Golgi apparatus, which is the next modification organelle. Thus, the ER functions as a production factory for proteins via rigid product quality controls to maintain cell homeostasis.
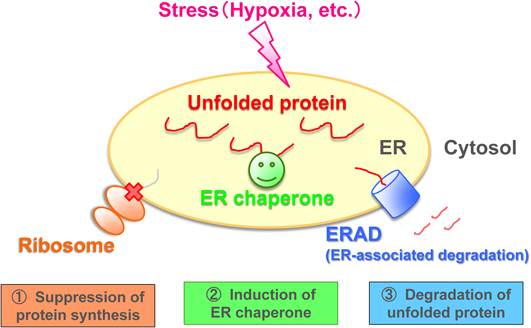
When a stressor blocks ER function (for example, hypoxia and glucose deprivation during ischemia, or protein overload from a virus), the ER overflows with immature defective proteins. This situation is called ER stress. If it continues over a sustained period, apoptosis signals are initiated from the ER, resulting in apoptotic cell death. To respond to ER stress, cells activate three defensive mechanisms, collectively called the unfolded protein response (UPR), aimed at reducing the defective proteins. The first defense mechanism is the arrest of further protein synthesis in the ribosomes. Second is the repair of defective proteins by ER-resident molecular chaperone mobilization. Third is the elimination and degradation of defective proteins from the ER1) (Fig. 1). The final mechanism is called ER-associated degradation (ERAD).2)
Initially, the UPR was analyzed for the molecular yeast mechanisms of inositol-requiring enzyme 1 (Ire1), which acts as an ER stress sensor and transduces information by transfer outside the ER. After mammalian Ire1 homologs (IRE1) were identified in 1998,3,4) considerable attention was then focused on the mammalian UPR. Later, two ER stress sensors that do not exist in yeast were identified,5,6) and at the same time the relationships of ER stress to various diseases were reported.7) ER stress and the UPR suddenly began to be highlighted. Beginning with a report in 1999 on the relationship between ER stress and Alzheimer’s disease (AD),8) relationships were subsequently found between ER stress and Parkinson’s disease (PD),9) polyglutamine disease,10) and other neurodegenerative diseases. Further relationships are indicated for many other diseases, including diabetes,11) metabolic syndrome,12) and cancer.13) Research related to ER stress-induced cellular dysfunction and disease pathogenesis has continued to expand explosively (Table 1). This review is a simple summary of the roles of ER stress in basic UPR and pathogenic mechanisms reported to date.
Table 1. ER Stress and Diseases
| Disease/Disorder | Cause | Literature |
|---|
| Alzheimer’s disease | •Mutant presenilin inhibits the UPR sensors | 8 |
| •Aβ induces ER stress | 29 |
| •HRD1 ubiquitinates and degrades APP | 30 |
| •Increase in S-nitrosylated PDI in patient brains | 30 |
| Parkinson’s disease | •Parkin mutation causes Pael-R accumulation in the ER | 9 |
| •MPTP induces ER stress | 31 |
| •Increase in S-nitrosylated PDI in patient brains | 32 |
| ALS | •Mutant SOD1 inhibits Derlin-1 | 33 |
| Diabetes | •Dysfunctional PERK and eIF2α induce β cell apoptosis | 11, 35, 36, 37 |
| •Insulin mutation induces ER stress in Akita mouse | 38 |
| •WFS1 mutation activates ATF6 by HRD1 destabilization | 41 |
| Metabolic syndrome | •ER stress induces steatosis | 42, 43 |
| •Chemical chaperone improves type 2 diabetes | 12, 44 |
| •Chemical chaperone suppresses adipocytic inflammation | 45 |
| Cancer | •Inhibition of ER stress sensors reduces tumorigenesis | 13, 47, 48, 49 |
Modified from Journal of Clinical and Experimental Medicine (IGAKU NO AYUMI), 254, 391–396 (2015) with permission.
2. ER STRESS SENSORS
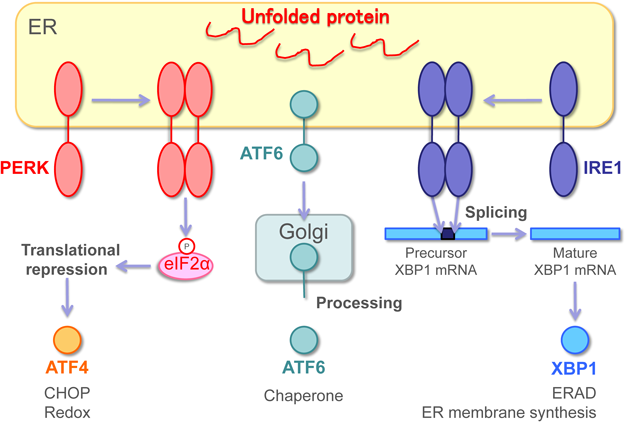
The primary mammalian ER stress sensors, in addition to IRE1, include activating transcription factor 6 (ATF6) and double-stranded RNA-dependent protein kinase (PKR)-like ER kinase (PERK) (Fig. 2). Only IRE1 are conserved from yeast, whereas ATF6 and PERK are newly added during the evolutionary process.
IRE1 has a serine/threonine kinase region and a ribonuclease (RNase) region on the cytoplasmic side (C-terminus). When ER stress is sensed, IRE1 forms dimers or oligomers, and is activated by autophosphorylation, which expresses RNase activity to process mRNA.3,4) The X-box binding protein 1 (XBP1) is a molecule in which IRE1 RNase activity mediates splicing to promote proper translation. The splicing by IRE1 differs from conventional splicing via spliceosomes; it is a non-conventional splicing for cutting matured mRNA.14,15) XBP1 is a transcription factor with a basic leucine zipper (bZIP) domain, and it binds to the promoter region of genes related to ER chaperones, ERAD, ER membrane synthesis, and protein secretion to promote their transcription.16,17)
ATF6 is a transcription factor of the cAMP response element-binding protein (CREB)/ATF family. In response to ER stress, ATF6 becomes free from the ER chaperone binding immunoglobulin protein (BiP), which is bound to the ATF6 ER luminal side (C-terminus).5,18) Furthermore, at steady state, ATF6 forms a disulfide bond in the ER lumen, whereas under ER stress conditions, it becomes a reduced form, into monomers that translocate to the Golgi apparatus.19) ATF6 undergoes two-step processing (called regulated intramembrane proteolysis, or RIP) by the site-1 protease (S1P) and site-2 protease (S2P) present in the Golgi, resulting in the release of the cytoplasm side (N-terminus) from the membrane.20) The separated ATF6 cytoplasmic side, including a bZIP domain, is translocated inside the nucleus and bound to the ER stress-response element (ERSE) to induce expression of the ER chaperone and other genes.5)
PERK is a kinase; it forms oligomers and is activated by autophosphorylation in response to ER stress, such as IRE1. The activated PERK phosphorylates the eukaryotic initiation factor 2α (eIF2α), which configures the ribosome to suppress protein translation.6) The PERK−eIF2α pathway acts, prior to improvement of ER stress conditions via the ATF6 and IRE1 pathways, to reduce the unfolded protein load on the ER by temporarily inhibiting protein synthesis. The eIF2α phosphorylation selectively promotes the translation of genes harboring an upstream open reading frame (uORF). The uORF is a reading frame that begins translation from 5′-upstream of the original mRNA coding region.21,22) There are a number of genes in which the original ORF or uORF is selected in response to cell differentiation, metabolism, and stress. Downstream of eIF2α, the activating transcription factor 4 (ATF4) is induced.23) During ER stress, ATF4, whose translation is promoted by uORF, performs the transcriptional induction of genes, CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP), a transcription factor implicated in apoptosis23) and is related to amino acid metabolism24) and oxidation/reduction.25)
3. ER-ASSOCIATED DEGRADATION (ERAD)
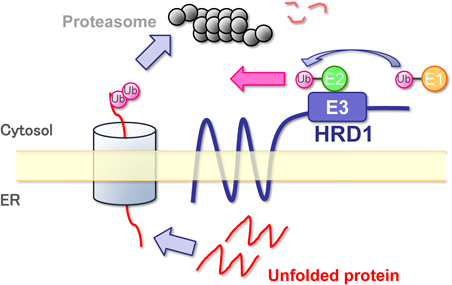
ERAD is a system for the degradation of proteins in the ER lumen and ER membrane through the ubiquitin−proteasome system in the cytoplasm; it is one of the defense mechanisms for ER stress that is conserved from yeast2) (Fig. 3). The eventual defective proteins, recognized by molecular chaperones, are discharged to the cytoplasm from the ER through a protein channel called the dislocon. The defective proteins undergo ubiquitination in the cytoplasm by the cooperative action of ubiquitin activating enzyme (E1), ubiquitin binding enzyme (E2), and ubiquitin ligase (E3). These poly-ubiquitinated proteins are quickly drawn from the ER membrane, and then recognized and degraded by proteasome.26) Of the enzymes involved in ubiquitination, the ubiquitin ligase plays the most important role in determining the recognition and degradation speed of a substrate protein. The classic HRD1 is an ER 5-transmembrane type ubiquitin ligase conserved from yeast, and its expression is induced by the UPR to regulate ERAD activity.27)
4. NEURODEGENERATIVE DISEASE
AD, PD, and amyotrophic lateral sclerosis (ALS) are well-known neurodegenerative diseases. Their pathogenic mechanisms remain unclear, and their fundamental therapeutic methods have not yet been established. A commonly observed pathological finding of neurodegenerative disease is the accumulation of denatured proteins. A relationship with ER stress has been reported in many neurodegenerative diseases.28)
In 1999, it was reported that mutations of presenilin, one of the genes causing familial AD, suppresses the activation of IRE1 and other ER stress sensors, resulting in attenuated UPR, and making neurons more vulnerable to ER stress.8) Subsequently, it was also determined that amyloid β (Aβ) deposited on the AD brain causes ER stress, thus contributing to cell toxicity.29) In addition, in another interesting report in understanding the pathology of AD, it was suggested that the ubiquitin ligase HRD1 involved in ERAD promotes ubiquitination and degradation of the amyloid precursor protein (APP), raising the possibility of reducing the Aβ level. Furthermore, the protein levels of HRD1 were significantly reduced in the cerebral cortex of AD patients.30)
Parkin (PARK2), one of the known genes that causes familial PD, is a ubiquitin ligase involved in ERAD. There is a hypothesis that parkin mutation causes the accumulation of a substrate, such as parkin-associated endothelin receptor-like receptor (Pael-R), in the ER, leading to ER stress, and resulting in ER stress-induced cell death of dopaminergic neurons in the substantia nigra.9) In addition, drugs which induce PD, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), cause ER stress, and the involvement of ATF6 activation in this process has been reported.31)
While nitric oxide (NO) is known to induce ER stress, protein-disulfide isomerase (PDI) that works on protein disulfide bond formation in the ER has been identified as a NO target. When cysteine residues in the PDI enzyme active sites are S-nitrosylated by NO, the enzyme activity declines, resulting in the accumulation of defective proteins in the ER. Interestingly, PDI S-nitrosylation increases in the post-mortem brain of AD and PD patients.32)
ALS is a neurodegenerative disease that damages the motor neurons. Cu/Zn-superoxide dismutase (SOD1), a causative gene for ALS, is localized in the cytoplasm. If its gene mutations cause changes in protein structure, it binds with Derlin-1, an ERAD-related molecule. As a consequence, the ERAD system is impaired, and ER stress is elicited.33)
In addition, since polyglutamine protein found in Huntington’s disease10) and prion proteins that cause Creutzfeldt-Jakob disease34) also induce ER stress, it would appear that many of the mutation proteins found in neurodegenerative diseases either directly or indirectly impair the ER function.
5. DIABETES
Diabetes has been indicated to have a relationship with ER stress as well. Insulin, a peptide hormone, undergoes disulfide bond formation and processing in the ER to exert ligand activity. In pancreatic β-cells that produce insulin, the ER is developed to respond to insulin production increased by as much as approximately 100-fold during secretion.
Knockout (KO) mice for the ER stress sensor PERK exhibited apoptosis in β-cells from around 15 d after birth, causing the onset of insulin-deficient Type 1 diabetes.11) Furthermore, even with PERK target eIF2α mutation (S51A)35) and PERK negative feedback factor P58IPK, which is induced downstream of PERK, KO mice36) exhibited a diabetic phenotype, suggesting that the PERK pathway plays an important role in the pathogenesis of diabetes. In Wolcott–Rallison syndrome, concomitantly presented with juvenile Type 1 diabetes and osteogenesis imperfecta, PERK gene mutation has also been found to be a cause.37)
In the Akita mouse presented with early-onset diabetes, a mutation in cysteine residue (C96Y) that is necessary for the formation of disulfide bonds has also been found to be a cause. This insulin mutation appears to form an unfolded structure that accumulates in the ER and induces apoptosis. When CHOP that induces apoptosis downstream of PERK was knocked out in the Akita mouse, the onset of diabetes was delayed.38) This therefore supports the idea that ER stress contributes to β-cell death in diabetes.
Pancreatic β-cell-specific Xbp1 conditional KO mice displayed impairment of proinsulin production and processing leading to decreased insulin secretion.39) Atf6α KO mice fed with a high fat diet also exhibited impaired proinsulin production. Pancreatic β-cells suffered from ER stress in the Atf6α-deficient mice.40) These findings suggest that the three major UPR pathways are involved in ER stress of pancreatic β-cells.
Wolfram syndrome is an autosomal recessive genetic disease that initially presents alongside juvenile diabetes mellitus, followed by complications of optic atrophy and diabetes insipidus. WFS1 has been identified as a causal gene. Wild-type WFS1 stabilizes HRD1 to promote the degradation of ATF6 and to negatively regulate the ATF6 signal. In Wolfram syndrome, the mutation of WFS1 appears to reduce the HRD1 function, and to induce excessive activation of the ATF6 pathway.41)
6. METABOLIC SYNDROME
While various environmental factors are involved in the pathogenesis of metabolic syndromes, recent attention has focused on ER stress. ER stress activates inflammation and various stress signals, and induces glucose and lipid metabolic disorders in β-cells, hepatocytes, and adipocytes. Thus, ER stress appears to induce obesity, insulin resistance, steatosis, dyslipidemia, and other metabolic syndromes.
ER stress promotes lipid synthesis and induces decreased lipoproteins, resulting in the development of steatosis. The overexpression of BiP in the liver of obese mice improves steatosis and promotes glucose utilization.42) In addition, if ER stress conditions continue, CHOP induction causes the suppression of the C/EBP α, which is an important transcription factor in lipid metabolism. As a result, this inhibits the oxidation of fatty acids, the secretion of lipoproteins, and gluconeogenesis, and promotes the development of steatosis.43)
Low molecular weight compound chemical chaperones are known to be ER stress inhibitors. Chemical chaperones prevent the aggregation of defective proteins in the ER, and promote protein transport from the ER to the target location. In the ob/ob mouse with Type 2 diabetes, application of the chemical chaperone 4-phenyl butyric acid (4-PBA) and tauroursodeoxycholic acid (TUDCA) improved insulin resistance and glycemic control.44)
High fat diet-fed mice exhibit chronic inflammation in adipose tissue. This inflammation response implicates ER stress mediated by reactive oxygen species and inflammatory cytokines due to free fatty acids. In mice fed a high fat diet, the administration of 4-PBA or TUDCA attenuates the UPR activation and inflammation response in liver and adipose tissue, suppresses weight gain, and improves insulin signaling.45) In other words, ER stress plays an important role in the dysfunction of adipose tissue associated with obesity. Indeed, subcutaneous adipose tissue of obese human subjects showed upregulation of several UPR stress-related proteins and activation of c-jun N-terminal kinase (JNK), which inhibits insulin action and activates proinflammatory pathways.46)
7. CANCER
The UPR is a mechanism activated to prevent apoptosis due to ER stress, whereas cancer abuses the UPR’s ability to acquire stress resistance and thus promotes cell proliferation. In solid cancers, angiogenesis cannot keep up with this rapid proliferation, leading to hypoxia and glucose deprivation in the internal microenvironment, and resulting in the generation of persistent ER stress. To overcome these ER stress conditions and to adapt to the poor environment for proliferation, cancer cells constitutively activate the UPR while suppressing apoptosis. In Perk-deficient mice, tumorigenicity and the expression of angiogenesis genes are attenuated.47) Meanwhile, since CHOP is expressed downstream of constitutively activated PERK by way of the transcription factor ATF4, this should induce apoptosis. However, cancers ingeniously escape from apoptosis because they suppress CHOP expression by inducing the expression of P58IPK that inhibits PERK activation.48) The IRE-XBP1 pathway is also important for cancer proliferation under hypoxic conditions. Xbp1-deficient mice exhibit increased cancer cell death as a result of hypoxia and decreased proliferation.13) ATF6 also appears to contribute to the survival and resistance of cancers in dormancy through mammalian target of rapamycin (mTOR) signaling.49) In conclusion, while cancer uses the UPR to survive and proliferate, suppressing the UPR in cancer patients may be an effective novel treatment method.
8. CONCLUSION
To date, attention has focused on ER stress as a cause of disease that results in damage to an organism, based on a literal “stress.” In recent years, however, functional analyses of ER stress sensors and their downstream genes have revealed that ER stress is positively involved in tissue differentiation and maturation. The UPR conserved from yeast was diversified in the process of evolution in multicellular organisms, and has developed as a variety of signaling pathways that affect cell differentiation and fate.50) In the future, if we are able to understand the complete picture of signaling pathways and physiological functions of UPR-related molecules, we can expect the elucidation of pathogenic mechanisms in diseases of unclear pathology, as well as the development of novel therapeutics.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol., 8, 519–529 (2007).
- 2) Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol., 3, 246–255 (2002).
- 3) Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev., 12, 1812–1824 (1998).
- 4) Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J., 17, 5708–5717 (1998).
- 5) Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem., 273, 33741–33749 (1998).
- 6) Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic–reticulum–resident kinase. Nature, 397, 271–274 (1999).
- 7) Yoshida H. ER stress and diseases. FEBS J., 274, 630–658 (2007).
- 8) Katayama T, Imaizumi K, Sato N, Miyoshi K, Kudo T, Hitomi J, Morihara T, Yoneda T, Gomi F, Mori Y, Nakano Y, Takeda J, Tsuda T, Itoyama Y, Murayama O, Takashima A, St George-Hyslop P, Takeda M, Tohyama M. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol., 1, 479–485 (1999).
- 9) Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell, 105, 891–902 (2001).
- 10) Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev., 16, 1345–1355 (2002).
- 11) Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell, 7, 1153–1163 (2001).
- 12) Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science, 306, 457–461 (2004).
- 13) Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res., 64, 5943–5947 (2004).
- 14) Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell, 107, 881–891 (2001).
- 15) Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature, 415, 92–96 (2002).
- 16) Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol., 23, 7448–7459 (2003).
- 17) Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell, 27, 53–66 (2007).
- 18) Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell, 10, 3787–3799 (1999).
- 19) Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol., 27, 1027–1043 (2007).
- 20) Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell, 6, 1355–1364 (2000).
- 21) Fernandez J, Yaman I, Merrick WC, Koromilas A, Wek RC, Sood R, Hensold J, Hatzoglou M. Regulation of internal ribosome entry site-mediated translation by eukaryotic initiation factor-2alpha phosphorylation and translation of a small upstream open reading frame. J. Biol. Chem., 277, 2050–2058 (2002).
- 22) Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem. Sci., 29, 152–158 (2004).
- 23) Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell, 6, 1099–1108 (2000).
- 24) Siu F, Bain PJ, LeBlanc-Chaffin R, Chen H, Kilberg MS. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J. Biol. Chem., 277, 24120–24127 (2002).
- 25) Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell, 11, 619–633 (2003).
- 26) Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol., 14, 19–57 (1998).
- 27) Kaneko M, Ishiguro M, Niinuma Y, Uesugi M, Nomura Y. Human HRD1 protects against ER stress-induced apoptosis through ER-associated degradation. FEBS Lett., 532, 147–152 (2002).
- 28) Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol., 12, 105–118 (2013).
- 29) Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature, 403, 98–103 (2000).
- 30) Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci., 30, 3924–3932 (2010).
- 31) Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem., 286, 7947–7957 (2011).
- 32) Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature, 441, 513–517 (2006).
- 33) Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev., 22, 1451–1464 (2008).
- 34) Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J., 22, 5435–5445 (2003).
- 35) Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell, 7, 1165–1176 (2001).
- 36) Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes, 54, 1074–1081 (2005).
- 37) Delépine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott–Rallison syndrome. Nat. Genet., 25, 406–409 (2000).
- 38) Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest., 109, 525–532 (2002).
- 39) Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A., 108, 8885–8890 (2011).
- 40) Usui M, Yamaguchi S, Tanji Y, Tominaga R, Ishigaki Y, Fukumoto M, Katagiri H, Mori K, Oka Y, Ishihara H. Atf6alpha-null mice are glucose intolerant due to pancreatic beta-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism, 61, 1118–1128 (2012).
- 41) Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Invest., 120, 744–755 (2010).
- 42) Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest., 119, 1201–1215 (2009).
- 43) Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, Katze MG, Hussain MM, Song B, Swathirajan J, Wang J, Yau GD, Kaufman RJ. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell, 15, 829–840 (2008).
- 44) Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science, 313, 1137–1140 (2006).
- 45) Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep., 2, 799 (2012).
- 46) Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes, 57, 2438–2444 (2008).
- 47) Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, Bell JC. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol., 26, 9517–9532 (2006).
- 48) Huber AL, Lebeau J, Guillaumot P, Petrilli V, Malek M, Chilloux J, Fauvet F, Payen L, Kfoury A, Renno T, Chevet E, Manie SN. p58(IPK)-mediated attenuation of the proapoptotic PERK–CHOP pathway allows malignant progression upon low glucose. Mol. Cell, 49, 1049–1059 (2013).
- 49) Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. U.S.A., 105, 10519–10524 (2008).
- 50) Saito A. Physiological functions of endoplasmic reticulum stress transducer OASIS in central nervous system. Anat. Sci. Int., 89, 11–20 (2014).