Abstract
Methylmercury (MeHg) results in cell death through endoplasmic reticulum (ER) stress. Previously, we reported that MeHg induces S-mercuration at cysteine 383 or 386 in protein disulfide isomerase (PDI), and this modification induces the loss of enzymatic activity. Because PDI is a key enzyme for the maturation of nascent protein harboring a disulfide bond, the disruption in PDI function by MeHg results in ER stress via the accumulation of misfolded proteins. However, the effects of MeHg on unfolded protein response (UPR) sensors and their signaling remain unclear. In the present study, we show that UPR is regulated by MeHg. We found that MeHg specifically attenuated inositol-requiring enzyme 1α (IRE1α)–x-box binding protein 1 (XBP1) branch, but not the protein kinase RNA-like endoplasmic reticulum kinase (PERK) and activating transcriptional factor 6 (ATF6) branches. Treatment with GSK2606414, a specific PERK inhibitor, significantly inhibited MeHg-induced cell death. These findings suggest that MeHg exquisitely regulates UPR signaling involved in cell death.
Methylmercury (MeHg), an environmental pollutant, is a neurotoxin that poses a major risk to human health.1) MeHg causes damage to the central nervous system, resulting in Minamata disease.2,3) MeHg has a high affinity for the sulfhydryl groups of proteins, facilitating its accumulation in various tissues.4) Particularly, MeHg is rapidly absorbed by the gastrointestinal tract and readily diffuses through the blood–brain barrier via the L-type large neutral amino acid transporter as MeHg–L-cysteine (Cys) conjugates.5) Therefore, the brain is a critical organ for MeHg toxicity. Also, MeHg toxicity is caused by binding of MeHg to the cysteinyl groups of proteins, which in turn interferes with the synthesis of cellular glutathione, thereby leading to oxidative damage.6) Alternatively, because MeHg has a high affinity for thiols, its toxicity is dependent on the modifications of covalent bonds (i.e., S-mercuration). Thus, the modifications involving the cysteine residues of intracellular proteins caused by MeHg result in the disruption of homeostasis or cell death.
Previously, we demonstrated that both nitric oxide (NO) and MeHg result in protein disulfide isomerase (PDI) dysfunction via covalent modifications involving the cysteine residues in active sites of proteins.7,8) S-Nitrosylation and -mercuration of PDI in active sites promote endoplasmic reticulum (ER) stress and unfolded protein response (UPR). We earlier demonstrated that inositol-requiring enzyme 1α (IRE1α), a sensor of ER stress that is located within the ER membrane, is also directly modified and regulated by NO.9) This modification leads to the inhibition of endonuclease activity, thereby attenuating the splicing of immature XBP1 mRNA. Because nucleophiles such as NO and MeHg easily react with protein thiols and affect enzymatic activity or function, it is important to determine its molecular targets and to characterize their effects on cells.10)
To date, the precise mechanism underlying MeHg-induced cell death remains elusive. Based on the findings of our previous study, we assume that MeHg also affects the condition of UPR sensors during ER stress, particularly in strongly inducing neuronal cell death. The aim of the present study was to elucidate the mechanism underlying cell death via a MeHg-induced ER stress/UPR.
MATERIALS AND METHODS
Materials and AntibodiesMeHg was purchased from Tokyo Chemical Industry (Tokyo, Japan). GSK2606414 was from Merck Millipore (Billerica, MA, U.S.A.). Antibodies against IRE1α, phospho-IRE1α, protein kinase RNA-like endoplasmic reticulum kinase (PERK), phospho-PERK, eukaryotic initiation factor 2 (eIF2) α, phospho-eIF2α, activating transcription factor 6 (ATF6) and β-actin were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.).
Cell CultureMouse embryonic fibroblasts (MEF) and human neuroblastoma SH-SY5Y cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10%(v/v) heat-inactivated fetal calf serum at 37°C in humidified atmosphere of 5% CO2/95% air.
Western Blot AnalysisCells were cultured in the indicated medium, harvested, washed with phosphate-buffered saline (PBS), and lysed in ice-cold lysis buffer [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM ethylenediamine N,N,N,′N′-tetraacetic acid ethylenediaminetetraacetic acid (EDTA), and 1% NP-40 containing a protease inhibitor cocktail] for 10 min.
To detect ATF6, the cells were pre-incubated with or without 50 µM of cycloheximide and 5 µM of MG-132 for 3 h and then further incubated for MeHg stimulation. After quantification by using the Bradford assay, the proteins were boiled in a sample loading buffer for 5 min and separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane and blocked with 5% non-fat dry milk or bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature. Then, the membrane was incubated with IRE1α (1 : 5000), phospho-IRE1α (1 : 5000), eIF2α (1 : 5000), phospho-eIF2α (1 : 5000), or β-actin (1 : 5000) antibodies in TBST and then detected by using respective anti-horseradish peroxidase (HRP)-linked secondary antibodies (1: 50000) in TBST. The antibody-reactive bands were visualized by enhanced chemiluminescence (ECL) using the ChemiDoc MP Imaging System (Bio-Rad). Blots were then quantified using ImageJ, and relative ratios were calculated (https://imagej.nih.gov/ij/docs/faqs.html).11,12)
RNA Extraction and RT-PCRTotal RNA was extracted using a TRI reagent. Transcriptor High Fidelity cDNA Synthesis Kit (Roche, Basel, Switzerland) was used to synthesize the cDNA according to the manufacturer’s instructions. The cDNA was amplified by using 25 or 30 cycles of PCR. The following primers were used: XBP1 sense primer, 5′-TTA CGA GAG AAA ACT CAT GGC C-3′; XBP1 antisense primer, 5′-GGG TCC AAG TTG TCC AGA ATG C-3′; β-actin sense primer, 5′-CCT GAC GGC CAG GTC ATC-3′; β-actin antisense primer, 5′-GGA CTC GTC ATA CTC CTG-3′. The PCR products were analyzed by agarose gel electrophoresis on a 1.5% gel.
Assessment of Nuclear MorphologyChromosomal condensation was assessed using the fluorescent dye Hoechst 33258. Briefly, cells were stained using 10 µM Hoechst33258 and washed with PBS. All samples were mounted and observed under a fluorescence microscope. The results were expressed as the percentage of cells with condensed nucleus versus the total number of cells.13)
Statistical AnalysisAll experiments were repeated independently at least three times. The results are expressed as the mean±standard error of the mean (S.E.M.) Statistical comparisons were performed by using ANOVA post hoc Bonferroni’s test (for multiple groups) using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, U.S.A.).
RESULTS AND DISCUSSION
Previous investigations have elucidated the mechanism of cell death in response to MeHg by inducing ER stress. However, the target molecules and the precise mechanism underlying MeHg-induced cell death remains unclear.14) We previously showed that PDI is a possible direct target of MeHg, and its modification leads to the attenuation of the enzymatic activity.8) The dysfunction of PDI results in ER stress and the activation of UPR by sensing the accumulation of immature unfolded proteins in ER lumen. In the present study, we examined the modulation of UPR via treatment with MeHg.
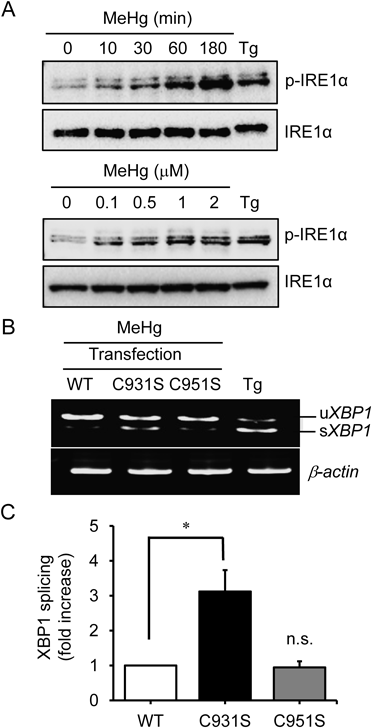
During ER stress, XBP1 mRNA is alternately spliced and activated by IRE1α, which is a UPR component.15) To address whether the IRE1α–XBP1 branch in UPR is affected by MeHg, we evaluated the process of IRE1α phosphorylation in MEF. Figure 1A shows that MeHg results in the phosphorylation of IRE1α in concentration- and time-dependent manners, which in turn suggests that MeHg induces ER stress and activates IRE1α by sensing the accumulation of misfolded proteins in the ER lumen. We previously found that NO elicited ER stress and then promoted IRE1α phosphorylation.9) Interestingly, the ribonuclease activity of IRE1α was strongly abolished via the S-nitrosylation of the kinase-extension nuclease (KEN) domain in IRE1α, although phosphorylated IRE1α was detected.16) Therefore, we consider that MeHg also regulates IRE1α endonuclease activity. Subsequently, we examined the splicing of immature XBP1 mRNA (a selective substrate) to assess the enzymatic activity of IRE1α. As expected, MeHg did not induce the cytosolic splicing of immature XBP1 mRNA in MEFs. The IRE1α–null MEFs were transfected with vectors encoding wild-type hemagglutinin (HA)-tagged IRE1α or cysteine mutants.17) MeHg-induced inhibition of IRE1α endonuclease activity was significantly ameliorated in the MEFs that expressed IRE1α (C931S), but not the IRE1α (C951S) mutants, thereby suggesting that C931 in IRE1α could be a predominant target of MeHg (Figs. 1B, C).
Next, we investigated whether other UPR branches were also modulated by MeHg. Treatment with MeHg stimulated the proteolysis of ATF6 (data not shown) and phosphorylation of PERK and eIF2α (Fig. 2A). A previous study has shown that the IRE1α–XBP1 branch functions as an anti-apoptotic pathway.18) In contrast, the PERK/ATF6 branches are involved in the induction of cell death.19) Therefore, these signals may be implicated in the MeHg-induced loss of cell viability. To determine this possibility, we tested the effect of a PERK-specific inhibitor (GSK2606414) on MeHg-induced cell death.20) Figure 2B shows that stimulation with MeHg triggers a striking change in cell and nuclear morphologies that were characterized by typical apoptotic features including nuclear condensation. Pre-incubation with GSK2606414 for 1 h significantly reduced the number of cells with condensed nucleus, which was a concentration-dependent response to MeHg (Fig. 2C). Apoptotic cell death was abolished by treatment with GSK2606414 in a dose-dependent manner, suggesting that the PERK pathway partially contributes to cell death.
The findings of the present study clearly show that MeHg induces cell death via ER stress. Although MeHg stimulates three major UPR sensors, IRE1α endonuclease activity and its downstream signaling are selectively attenuated by the interaction with or modulation of cysteine 931 in the KEN domain of IRE1α. On the other hand, a pharmacological study using GSK2606414 determined that PERK is implicated in MeHg-induced cell death. Here, we report that MeHg disrupts anti-apoptotic signaling based on the IRE1α–XBP1 branch and simultaneously promotes pro-apoptotic signaling via the PERK/ATF6 branches. Therefore, the MeHg-induced promotion of pro-apoptotic signaling that is associated with CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) expression may be the predominant mechanism underlying cell death. Several electrophiles that can covalently modify the cysteine residues in proteins may modulate or regulate UPR signaling, particularly IRE1α. Taken together, in conjunction with the MeHg-induced attenuation of the IRE1α component of UPR, the activation of PERK may further sensitize cells to apoptotic death. Furthermore, these findings may be utilized in the development of novel therapeutic approaches for Minamata disease.
Acknowledgments
We thank Ms. Yoko Okamoto for providing technical assistance. The Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan Grants-in-Aid for Scientific Research (B) 15H04649, Challenging Exploratory Research 15K14952; The Shimabara Science Promotion Foundation; and the Smoking Research Foundation supported this study.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Järup L. Hazards of heavy metal contamination. Br. Med. Bull., 68, 167–182 (2003).
- 2) Harada M. Minamata Disease: Methylmercury poisoning in Japan caused by environmental pollution. Crit. Rev. Toxicol., 25, 1–24 (1995).
- 3) Eto K. Minamata disease. Neuropathology, 20 (Suppl.), S14–S19 (2000).
- 4) Bahr GF, Moberger G. Methyl-mercury-chloride as a specific reagent for protein-bound sulfhydryl groups: Electron stains II. Exp. Cell Res., 6, 506–518 (1954).
- 5) Kerper LE, Ballatori N, Clarkson TW. Methylmercury transport across the blood–brain barrier by an amino acid carrier. Am. J. Physiol., 262, R761–R765 (1992).
- 6) Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, Bainy A, Marques MR, Dafre AL, Farina M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic. Biol. Med., 47, 449–457 (2009).
- 7) Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-Nitrosylated protein-disulfide isomerase links protein misfolding to neurodegeneration. Nature, 441, 513–517 (2006).
- 8) Makino K, Okuda K, Sugino E, Nishiya T, Toyama T, Iwawaki T, Fujimura M, Kumagai Y, Uehara T. Correlation between attenuation of protein disulfide isomerase activity through S-mercuration and neurotoxicity induced by methylmercury. Neurotox. Res., 27, 99–105 (2015).
- 9) Nakato R, Ohkubo Y, Konishi A, Shibata M, Kaneko Y, Iwawaki T, Nakamura T, Lipton SA, Uehara T. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep., 5, 14812 (2015).
- 10) Unoki T, Abiko Y, Toyama T, Uehara T, Tsuboi K, Nishida M, Kaji T, Kumagai Y. Methylmercury, an environmental electrophile capable of activation and disruption of the Akt/CREB/Bcl-2 signal transduction pathway in SH-SY5Y cells. Sci. Rep., 6, 28944 (2016).
- 11) Numajiri N, Takasawa K, Nishiya T, Tanaka H, Ohno K, Hayakawa W, Asada M, Matsuda H, Azumi K, Kamata H, Nakamura T, Hara H, Minami M, Lipton SA, Uehara T. On-off system for PI3-kinase–Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc. Natl. Acad. Sci. U.S.A., 108, 10349–10354 (2011).
- 12) Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675 (2012).
- 13) Tsuruma K, Nakagawa T, Morimoto N, Minami M, Hara H, Uehara T, Nomura Y. Glucocorticoid modulatory element-binding protein 1 binds to initiator procaspases and inhibits ischemia-induced apoptosis and neuronal injury. J. Biol. Chem., 281, 11397–11404 (2006).
- 14) Usuki F, Fujita E, Sasagawa N. Methylmercury activates ASK1/JNK signaling pathways, leading to apoptosis due to both mitochondria- and endoplasmic reticulum (ER)-generated processes in myogenic cell lines. Neurotoxicology, 29, 22–30 (2008).
- 15) Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol., 8, 519–529 (2007).
- 16) Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J., 30, 894–905 (2011).
- 17) Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res., 315, 2496–2504 (2009).
- 18) Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science, 318, 944–949 (2007).
- 19) Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J., 283, 2640–2652 (2016).
- 20) Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem., 55, 7193–7207 (2012).