Abstract
Panaxydol, a polyacetylenic compound derived from Panax ginseng has been reported to suppress the growth of cancer cells. However, the molecular mechanisms underlying cell cycle arrest by this compound in non-small cell lung cancer (NSCLC) are unknown. Our study found that panaxydol treatment induced cell cycle arrest at G1 phase in NSCLC cells. The cell cycle arrest was accompanied by down-regulation of the protein expression of cyclin-dependent kinase (CDK) 2, CDK4, CDK6, cyclin D1 and cyclin E, and decrease in the phosphorylation of retinoblastoma (Rb) protein. Furthermore, up-regulation of cyclin-dependent kinase inhibitor (CDKI) p21CIP1/WAF1 and p27KIP1 was observed in panaxydol-treated NSCLC cells. In addition, panaxydol also induced accumulation of intracellular Ca2+ ([Ca2+]i). (Acetyloxy)methyl 2-({2-[(acetyloxy)methoxy]-2-oxoethyl}[2-(2-{2-[bis({2-[(acetyloxy)methoxy]-2-oxoethyl})amino]phenoxy}ethoxy)phenyl]amino)acetate (BAPTA-AM), the Ca2+ chelator, attenuated not only panaxydol-induced accumulation of [Ca2+]i, but also G1 cell cycle arrest and decrease of CDK6 and cyclin D1 protein expression level. These results demonstrated that the anti-proliferative effects of panaxydol were caused by cell cycle arrest, which is closely linked to the up-regulation of [Ca2+]i and represents a promising approach for the treatment of lung cancer.
Lung cancer is one of the leading causes of death from cancer worldwide and is the most common cause of cancer-associated death in the United States.1,2) Among them, 80–85% of cases diagnosed with lung cancer are classified as non-small cell lung cancer (NSCLC)3) which has a 5-year prognosis less than 15%.4) Screening of compounds, which show tumor-suppressing effects in NSCLC is important for the discovery of advanced treatments for NSCLC.
Most cells possess a ubiquitous and complex process known as cell cycle involving cell growth, proliferation and repair of DNA damage.5) The cell cycle is separated into four phases: DNA replication occurs in S phase, and division into two daughter cells occurs in M phase. G1 phase following mitosis involves response to growth signalling network. G2 phase follows S phase and indicates the time for mitosis or cell division.5) Cell-cycle progression is regulated by several cyclin-dependent kinases (CDK) and cyclin proteins.6) Cyclin D-CDK4/6 complex and cyclin E-CDK2 complex regulate progression from G1 to S phase via hypophosphorylation of retinoblastoma (Rb) protein and activation of E2F, which acts as a transcriptional factor for DNA synthesis.7) In cancer, gene mutations encoding several proteins, which are important for cell cycle result in abnormal and unrestrained cell proliferation. In this regard, targeting the cell cycle in cancer cells has been suggested as an alternative therapeutic strategy for drug discovery.8)
Natural products have been evaluated as the most important candidates among therapeutic drugs and many compounds derived from natural product have been approved as anti-cancer agents since 1940s.9) As a well-known natural product in Asia, Panax ginseng C.A. MEYER has been used traditionally, and many studies have investigated the anti-tumor activity of compounds derived from ginseng in vitro and in vivo.10) Among these compounds, panaxydol (heptadeca-1-en-4,6-diyn-9,10-epoxy-3-ol), a polyacetylenic compound isolated from Panax ginseng C.A. MEYER has been reported to suppresses cell proliferation via induction of p27KIP in human melanoma cell line and rat C6 glioma cell,11–13) and to induce apoptosis through epidermal growth factor receptor (EGFR) activation, endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) and mitogen-activated protein kinase (MAPK) pathway.14,15) However, the molecular mechanism between panaxydol-induced cell cycle arrest and intracellular Ca2+ level in non-small lung cancer cell has yet to be reported. In this regard, we investigated the mechanism underlying the anti-proliferative activities of panaxydol in NSCLC cells, A549 and NCI-H358.
MATERIALS AND METHODS
MaterialsPanaxydol was obtained from Chromadex (Irvine, CA, U.S.A.). RPMI 1640 medium, fetal bovine serum (FBS), penicillin and streptomycin sulfate were purchased from Life Technologies Inc. (Grand Island, NY, U.S.A.). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI), bisacrylamide, sodium dodecyl sulfate (SDS), phenylmethylsulfonyl fluoride (PMSF), (acetyloxy)methyl 2-({2-[(acetyloxy)methoxy]-2-oxoethyl}[2-(2-{2-[bis({2-[(acetyloxy)methoxy]-2-oxoethyl})amino]phenoxy}ethoxy)phenyl]amino)acetate (BAPTA-AM) and other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Ribonuclease A (RNase A) and tetramethylethylenediamine (TEMED) were purchased from Bio-Rad Laboratories (Portland, ME, U.S.A). Antibodies against CDK2, CDK4, CDK6, cyclin D1, cyclin E, p21CIP1/WAF1, p27KIP1, total Rb and β-actin were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, U.S.A.). p-Rb antibody was purchased from Cell Signaling Technology (Danvers, MA, U.S.A).
Cell CultureHuman adenocarcinomic alveolar basal epithelial A549 cells and human bronchioalveolar carcinoma NCI-H358 cells were obtained from the Korean cell line bank (Seoul, Korea). Cells were cultured in RPMI 1640 supplemented with 10% heated-inactivated FBS, penicillin (100 units/mL) and streptomycin sulfate (100 µg/mL). All cells were cultured under atmosphere of 5% CO2 at 37°C.
MTT AssayThe cells were seeded in 96-well plate, and each well contains 100000 cells/mL in 100 µL of medium supplemented with 10% FBS. The plates were incubated for 24 h, and then various concentrations (200, 100, 50, 25, 12.5, and 6.25 µM) of panaxydol were added in triplicate. After treatment for 48 h, 20 µL of MTT solution (5 mg/mL stock solution in phosphate buffered saline (PBS)) was consecutively treated and the cells were incubated for additional 4 h in dark. The medium was removed and formazan blue formed by cells was dissolved with 200 µL dimethyl sulfoxide (DMSO). Optical density was measured by microplate reader at 540 nm.
Cell Cycle AnalysisAfter treatment with different concentrations of panaxydol for various times, the cells were harvested with trypsin and washed twice with ice-cold PBS. The pellets were resuspended and fixed in 70% ethanol at 4°C for overnight. Before detected by flow cytometry, the cells were washed twice with PBS and resuspended in PI solution containing PI (50 mg/mL) and RNase A (250 mg/mL) for 30 min. The cells were analyzed by FACS cytometer.
Western Blot AnalysisAfter treatment of various concentration of panaxydol for different times, the cells were harvested and centrifuged at 2500×g for 7 min. And then washed twice with ice-cold PBS, then centrifuged at 21100×g for 5 min. The cell pellet was resuspended in protein lysis buffer (Intron, Seoul, Korea) for 30 min in ice. Cell debris was removed by microcentrifuge at 21100×g for 30 min and then the protein concentration of supernatants was determined by Biorad protein assay reagent, according to the manufacturer’s instruction. Thirty micrograms of protein was equally loaded and separated by SDS-PAGE on 8–10% polyacrylamide gel, and transferred onto polyvinylidene difluoride (PVDF), which incubated for 1 h with blocking solution at room temperature, and then with primary antibody at 4°C for overnight. Blots were washed three times with Tween 20/Tris-buffered saline (T/TBS) and incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature and rewashed three times with T/TBS and detected using enhanced chemiluminescence (Amersham, Buckinghamshire, U.K.) detection system.
Measurement of the Intracellular Calcium ([Ca2+]i)[Ca2+]i was measured by using cell membrane permeable fluorescent dye, Fluo-4/AM assay kit (Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer’s instructions. Briefly, cells were seeded 96-well plate and after 24 h and changed with assay buffer (1×HBSS, 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 2.5 mM probenecid). The free cytosolic Ca2+ levels were then assessed by fluorometer detecting Fluo-4/AM, Ca2+-indicator dye.
Statistical AnalysisAll values were expressed as the mean±standard deviation (S.D.) of triplicate experiments. Statistically significant values were compared using ANOVA and Dunnett’s post hoc test, and p-values of less than 0.05 were considered statistically significant.
RESULTS AND DISCUSSION
Panaxydol Suppresses the Proliferation in NSCLC CellsTo investigate whether panaxydol (Fig. 1A) has anti-proliferative activity against NSCLC cells, the MTT assay was performed to evaluate the cytotoxicity in cell lines derived from non-small cell lung cancer (Fig. 1B). As shown in Fig. 1B, viability of both cells was decreased in a dose-dependent manner, with IC50 values of 81.89 µM and >200 µM in A549 and NCI-H358 cells, respectively. It suggested that panaxydol reduces the growth of NSCLC cells, and the cytotoxic effect was stronger in A549 cells than in NCI-H358 cells.
Panaxydol Induces Cell Cycle Arrest at G1 Phase in NSCLC CellsCell cycle arrest is an important factor in the inhibition of tumor progression, which leads to cell-cycle phase transition regulated by various factors. To identify the anti-proliferative activity of panaxydol against NSCLC cells, cell cycle analysis was carried out in A549 and NCI-H358 cells using fluorescence-activated cell sorting (FACS) cytometry. When cells were treated with various concentrations (10, 20, and 30 µM in A549 and 10, 30, and 50 µM in NCI-H358 cells) of panaxydol, cell-cycle arrest occurred in G1 phase accompanying a decrease in S and G2/M phases when compared with untreated control cells. The proportion of G1 phase was increased from 65.1 to 85.8% in A549 cells and from 57.5 to 70.3% in NCI-H358 cells after treatment with various doses of panaxydol for 24 h, respectively (Fig. 2A). In addition, the proportion of G1 phase increased from 63.9 to 82.4% and from 60.1 to 66.6% after treatment with 30 µM and 50 µM panaxydol for different durations (Fig. 2B).
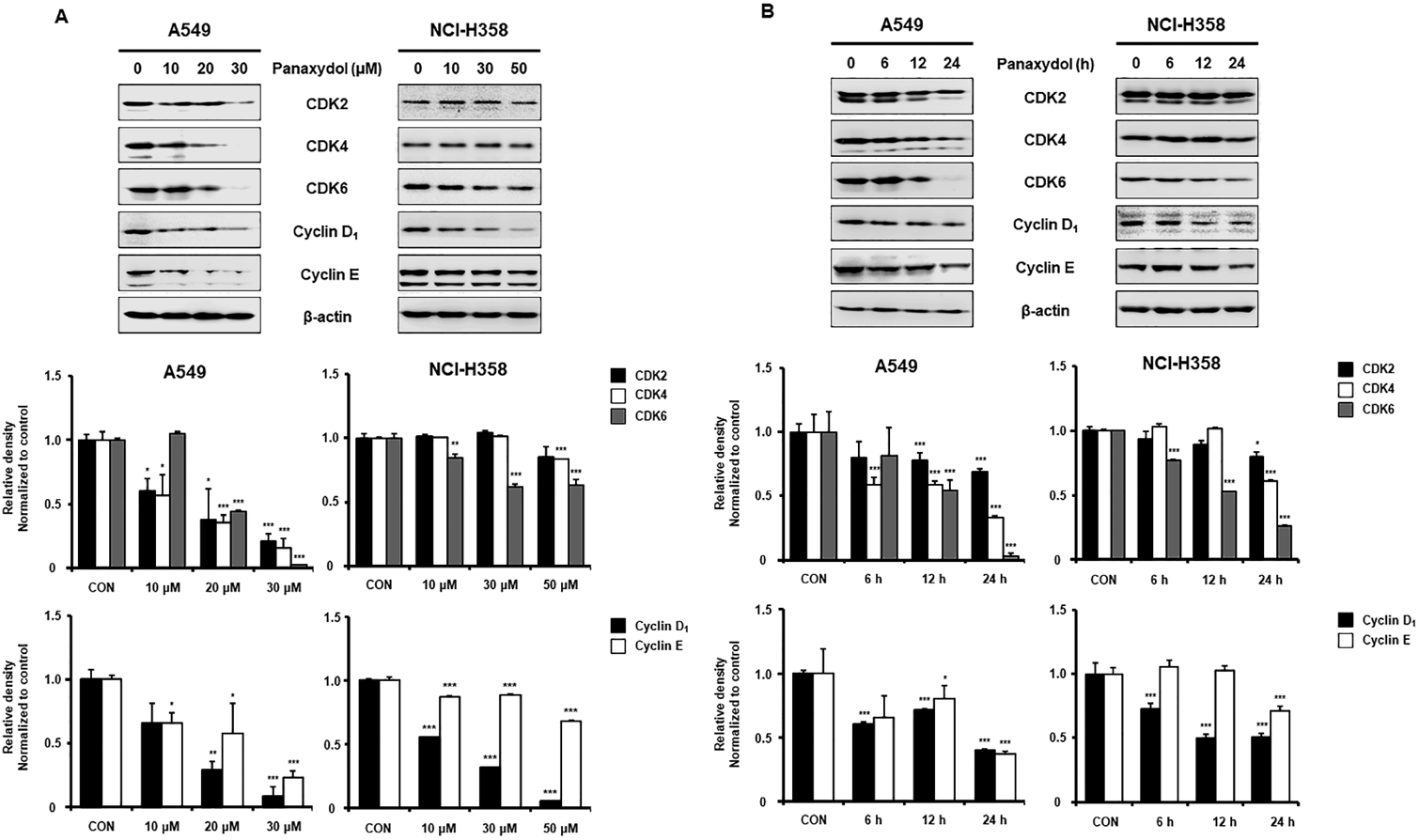
Panaxydol Downregulates Expression of G1 Phase Regulatory Proteins Including CDK–Cyclin Complex Protein in NSCLC CellsCell cycle progression is tightly regulated by several factors called cyclins associated with CDKs.16) In particular, cyclin D and E, which accompany CDK4, CDK6 and CDK2 are responsible for G1 phase progression.17) In this regard, we examined the expression of G1 phase-related proteins in panaxydol-treated cells. In A549 and NCI-H358 cells, panaxydol reduced the protein level of CDK2, 4, 6, cyclin D1 and cyclin E in a time- and dose-dependent manner (Figs. 3A, B). These results suggested that panaxydol induced G1 cycle arrest via downregulation of CDK2, 4, 6, cyclin D1 and cyclin E, which play a vital role in the progression of G1 cell cycle.
Panaxydol Inhibits Rb Phosphorylation and the Protein Expression of Cyclin-Dependent Kinase Inhibitors in NSCLC CellsG1 phase is triggered by the phosphorylation of Rb by G1 phase-related CDKs and cyclins, which are negatively regulated by CDK inhibitors (CDKI)18) resulting in the release of E2F, which initiates transcription for G1-S transition.19) Among the CDKIs, Cip/Kip family inactivates the CDK–cyclin complex.8,20) Moon et al. reported that panaxydol induced G1 phase cell cycle arrest caused by interaction of p27KIP1 with CDK2 in melanoma cells.13) Similarly, in our study, Western blot analysis revealed that panaxydol downregulated the phosphorylation of Rb without any effect on the expression of total Rb (Figs. 4A, B). In addition, increase of p21CIP1/WAF1 and p27KIP1 expression induced by panaxydol was confirmed in A549 and NCI-H358 cells. These results suggested that panaxydol induced G1 cell cycle arrest via suppression of CDK–cyclin protein expression and phosphorylation of Rb, as well as the ascent of p21CIP1/WAF1 and p27KIP1expression in NSCLC cells.
Panaxydol-Induced G1 Cell Cycle Arrest Is Attenuated by BAPTA-AM in NSCLC Cell LinesAs shown in Fig. 5A, panaxydol increase [Ca2+]i immediately after treatment, was shown in both NSCLC cells. In addition, pretreatment with BAPTA-AM, a cell-permeant chelator, prevented calcium level from increasing in an hour, compared with treatment with panaxydol only (Fig. 5B). This result corresponds to which panaxydol-induced G1 arrest was recovered by BAPTA-AM (Fig. 5C), and this effect also appeared in expression of cyclin D1 and CDK6 which known as regulatory protein of G1 phase in A549 cells (Fig. 5D). Although we found that BAPTA-AM attenuated panaxydol-induced decrease of cyclin D1 in a dose-dependent manner, we did not find any recovery effect of BAPTA-AM on panaxydol-induced CDK6 expression in NCI-H358 (Fig. 5D). In these regards, we speculated that panaxydol-regulated Ca2+ increase could affect to the formation of complex between cyclins and CDKs in NCI-H358 cells and further studies are need to reveal these suppositions. Based on these results, cell cycle arrest by panaxydol is related with change of [Ca2+]i accompanied with decrease in protein level of CDK–cyclin complex in A549 and NCI-H358 cells.
Overload of Ca2+ influx into cancer cells leads to dysfunction of cell cycle progression,21) and this process is possibly initiated by [Ca2+]i signaling. A variety of channels and pumps located on plasma membrane or ER coordinately control Ca2+ homeostasis of cytoplasm or ER. Because cells maintain a low Ca2+ background in the cytoplasm with concentrations of ca. 100 nm, Ca2+ signals are generated due to Ca2+ influx from the extracellular space with concentration around 1–2 mM, or Ca2+ release from intracellular Ca2+ stores, primarily the ER with concentrations of 250–600 µM.22) Accordingly, a previous study of panaxydol proved that panaxydol-induced activation of PLC-γ following EGFR activation triggers enhance of Ca2+ level in ER and provided panaxydol can induce Ca2+ increase through ER.15) Although the effect of panaxydol to Ca2+ level results in activation of apoptosis in human cervical cancer cell, it has been reported that prolonged arrest of cancer cells caused cell death.23–25) Furthermore, human cancer cells growing in vitro either enter into a stable arrest or die, depending on the integrity of their cell-cycle checkpoints during anti-cancer therapy.26) With these reports, our results suggest the possibility that panaxydol-induced [Ca2+]i increase affect cell proliferation differently, resulting in cell cycle arrest with down regulation of CDKs and cyclins rather than apoptosis. Moreover, our data has shown that panaxydol-induced downregulation of cell cycle associated protein was restored by Ca2+ chelator. Finding a molecular mechanism how increase of [Ca2+]i affect CDK–cyclin proteins is not elucidated yet, but we can make a supposition that panaxydol-generated [Ca2+]i increase stimulates Ca2+ sensors, such as calmodulin, and they are known to transmit signals eliciting downstream responses27) and contribute to control of cell cycle progression.28) Based on these findings, we suggest that panaxydol controls cell cycle phase induced by [Ca2+]i imbalance in lung cancer cells and further studies are needed to prove this supposition.
In conclusion, panaxydol attenuates cell cycle progression at G1 phase via regulation of G1-mediated protein expression, and its effects are attributed to enhancing [Ca2+]i in NSCLC cells. Our findings provide molecular evidence supporting the chemotherapeutic potential of panaxydol in lung cancer, particularly NSCLC.
Acknowledgments
This work was supported by the research Grant from the Korea Food Research Institute (E0145202).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv. Exp. Med. Biol., 893, 1–19 (2016).
- 2) Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J. Clin., 67, 7–30 (2017).
- 3) Petersen I, Petersen S. Towards a genetic-based classification of human lung cancer. Anal. Cell. Pathol., 22, 111–121 (2001).
- 4) Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J. Clin., 56, 106–130 (2006).
- 5) Williams GH, Stoeber K. The cell cycle and cancer. J. Pathol., 226, 352–364 (2012).
- 6) Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512 (1999).
- 7) Stewart ZA, Westfall MD, Pietenpol JA. Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol. Sci., 24, 139–145 (2003).
- 8) Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif., 36, 131–149 (2003).
- 9) Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J. Ethnopharmacol., 100, 72–79 (2005).
- 10) Chang YS, Seo EK, Gyllenhaal C, Block KI. Panax ginseng: a role in cancer therapy? Integr. Cancer Ther., 2, 13–33 (2003).
- 11) Guo L, Song L, Wang Z, Zhao W, Mao W, Yin M. Panaxydol inhibits the proliferation and induces the differentiation of human hepatocarcinoma cell line HepG2. Chem. Biol. Interact., 181, 138–143 (2009).
- 12) Hai J, Lin Q, Lu Y, Yi J, Zhang H. Growth inhibition and induction of differentiation by panaxydol in rat C6 glioma cells. Neurol. Res., 30, 99–105 (2008).
- 13) Moon J, Yu SJ, Kim HS, Sohn J. Induction of G(1) cell cycle arrest and p27(KIP1) increase by panaxydol isolated from Panax ginseng. Biochem. Pharmacol., 59, 1109–1116 (2000).
- 14) Kim JY, Yu SJ, Oh HJ, Lee JY, Kim Y, Sohn J. Panaxydol induces apoptosis through an increased intracellular calcium level, activation of JNK and p38 MAPK and NADPH oxidase-dependent generation of reactive oxygen species. Apoptosis, 16, 347–358 (2011).
- 15) Kim HS, Lim JM, Kim JY, Kim Y, Park S, Sohn J. Panaxydol, a component of Panax ginseng, induces apoptosis in cancer cells through EGFR activation and ER stress and inhibits tumor growth in mouse models. Int. J. Cancer, 138, 1432–1441 (2016).
- 16) Casimiro MC, Crosariol M, Loro E, Li Z, Pestell RG. Cyclins and cell cycle control in cancer and disease. Genes Cancer, 3, 649–657 (2012).
- 17) Donjerkovic D, Scott DW. Regulation of the G1 phase of the mammalian cell cycle. Cell Res., 10, 1–16 (2000).
- 18) Alexander K, Hinds PW. Requirement for p27(KIP1) in retinoblastoma protein-mediated senescence. Mol. Cell. Biol., 21, 3616–3631 (2001).
- 19) Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer, 1, 1124–1131 (2010).
- 20) Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med., 10, e19 (2008).
- 21) Parkash J, Asotra K. Calcium wave signaling in cancer cells. Life Sci., 87, 587–595 (2010).
- 22) Machaca K. Ca(2+) signaling, genes and the cell cycle. Cell Calcium, 48, 243–250 (2010).
- 23) Borel F, Lacroix FB, Margolis RL. Prolonged arrest of mammalian cells at the G1/S boundary results in permanent S phase stasis. J. Cell Sci., 115, 2829–2838 (2002).
- 24) Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell, 23, 567–576 (2012).
- 25) Hain KO, Colin DJ, Rastogi S, Allan LA, Clarke PR. Prolonged mitotic arrest induces a caspase-dependent DNA damage response at telomeres that determines cell survival. Sci. Rep., 6, 26766 (2016).
- 26) Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J. Cell-cycle arrest versus cell death in cancer therapy. Nat. Med., 3, 1034–1036 (1997).
- 27) Kudla J, Batistic O, Hashimoto K. Calcium signals: the lead currency of plant information processing. Plant Cell, 22, 541–563 (2010).
- 28) Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev., 24, 719–736 (2003).