Abstract
The purpose of this research was to establish an in vitro dissolution testing method to predict the oral pharmacokinetic (PK) profiles and food effects of gabapentin enacarbil formulated as wax matrix extended-release (ER) tablets in humans. We adopted various biorelevant dissolution methods using the United States Pharmacopeia (USP) apparatus 2, 3 and 4 under simulated fasted and fed states. Simulated PK profiles using the convolution approach were compared to published in vivo human PK data. USP apparatus 2 and 4 underestimated the in vivo performance due to slow in vitro dissolution behaviors. In contrast, biorelevant dissolution using USP apparatus 3 coupled with the convolution approach successfully predicted the oral PK profile of gabapentin enacarbil after oral administration of a Regnite® tablet under fasted state. This approach might be useful for predicting the oral PK profiles of other drugs formulated as wax matrix-type ER tablets under fasted state.
Oral extended-release (ER) formulations of drugs are developed to reduce side effects and maximize drug efficacy by modifying their pharmacokinetic (PK) characteristics. The formulation of a drug is not always optimized in the early phase of clinical trials. Once it is decided that a drug will be developed as an ER formulation, however, extensive formulation work is needed to establish the manufacturability and stability of the drug product. Possible changes in the PK of dosage forms throughout the course of clinical development are taken into consideration during ER formulation development. If the PK changes significantly, reformulation and additional clinical trials are required because the PK profile of a drug can affect its pharmacological effect and/or safety profiles. Therefore, exploratory clinical studies are sometimes implemented to understand the relationship between the rate of drug release and the PK of the drug. Accordingly, prediction of the in vivo performance of ER formulations in advance can reduce the development period with minimization of clinical trials.
In recent years, a number of technologies have been developed to enable prediction of the in vivo performance of dosage forms. For oral immediate release (IR) dosage forms, many researchers have successfully used biorelevant dissolution testing coupled with physiologically-based pharmacokinetic modeling approaches, to predict in vivo drug performance.1–4) Okumu et al.5) established the in vitro/in vivo correlation (IVIVC) for etoricoxib solid oral drug products by comparing the dissolution behavior in different dissolution media and using computer simulations.
Methods developed to predict the oral performance of ER dosage forms have also been reported. Klein et al.6) established the Level A IVIVC for two ER formulations containing caffeine using the United States Pharmacopeia (USP) apparatus 3 (reciprocating cylinder, BioDis) and 4 (flow-through cell). Fotaki et al.7) reported the importance of in vitro hydrodynamics when using the dissolution apparatus 2 (paddle), 3 and 4 to determine the IVIVC of two monolithic ER products, a hydrophilic matrix formulation containing carbomer and an osmotic pump formulation. Andreas et al.8) developed in vitro biorelevant test setups by using the USP dissolution apparatus 3 and 4 to predict the food effects of several ER formulations of 5-aminosalicylic acid which were pH-dependent coating formulations, a Multi-Matrix system, and ethylcellulose-coated microgranules. Further, Andreas et al.9,10) predicted the food effects of two nifedipine ER formulations (an osmotic pump and a matrix-type coat-core) and ER zolpidem (Ambien® CR). Garbacz et al.11,12) developed a dissolution stress test device to simulate the physiologically-based mechanical stress that a dosage form may experience when it moves through the gastrointestinal (GI) tract, and evaluated the performance of ER tablets with hydrophilic matrices containing 100 mg diclofenac sodium.
Gabapentin enacarbil is a prodrug of gabapentin that is effective in the treatment of restless legs syndrome and has been developed as a wax matrix, erosion-based ER tablet comprising glycerin fatty acid ester.13) While many attempts have been made to predict the in vivo performance of oral ER formulations, as mentioned above, we are unaware of any efforts to predict the in vivo performance of wax matrix-type oral ER formulations.
The purpose of this research was to establish an in vitro dissolution method to predict the oral PK profiles and food effects of gabapentin enacarbil ER tablets. By combing in vitro dissolution profiles of the ER tablet in simulated fasted and fed biorelevant media with a convolution approach using post-absorptive PK parameters in humans, we compared the predicted in vivo plasma concentration profiles of gabapentin with observed data under both prandial states.
MATERIALS AND METHODS
MaterialsCommercially available gabapentin enacarbil ER tablets 300 mg (Regnite® tablets 300 mg, Astellas Pharma Inc., Tokyo, Japan, lot L001) were purchased from the Japanese market. Gabapentin enacarbil ER tablets are a wax matrix-type formulation containing glycerin fatty acid ester, with a tablet size of 15.1×8.0 mm. Gabapentin enacarbil powder was produced by Astellas Pharma Inc. (lot 1001140G). FaSSIF/FeSSIF/FaSSGF powder (lot 01-1504-05NP) and FaSSIF-V2 powder (lot 02-1405-09) were purchased from Biorelevant.com Ltd. (London, U.K.). Acetic acid, acetonitrile, glycerol monooleate, hydrochloric acid solution (1 mol/L HCl), maleic anhydrate, phosphoric acid, potassium dihydrogen phosphate, sodium acetate, sodium chloride, sodium hydroxide pellets and sodium hydroxide solution (1 mol/L NaOH) were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). D(+)-Glucose was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Tris-(hydroxymethyl) aminomethane was purchased from MP Biomedicals, LLC. (Santa Ana, CA, U.S.A.). Milk (Meiji Holdings Co., Ltd., Tokyo, Japan) containing 3.5% fat was purchased commercially. Pepsin (lot SLBB2141V), sodium cholate (lot SLBN0541V), sodium oleate (lot SLBM1481V) and sodium taurocholate (lot SLBD5492V) were purchased from Sigma-Aldrich Co., LLC. (St. Louis, MO, U.S.A.). Lecithin (Lipoid E PC S, lot 510800-2140089-04/046) was purchased from Lipoid GmbH (Ludwigshafen, Germany).
Dissolution MediaThe following biorelevant dissolution media were used for solubility and dissolution testing: Fasted State Simulated Gastric Fluid (FaSSGF), Fed State Simulated Gastric Fluid (FeSSGFmiddle), Fasted State Simulated Intestinal Fluid version 2 (FaSSIF-V2), Fed State Simulated Intestinal Fluid version 2 (FeSSIF-V2), Fasted State Simulated Intestinal Fluid at midgut (FaSSIFmidgut), Fed State Simulated Intestinal Fluid at midgut (FeSSIFmidgut), Simulated Intestinal Fluid in the ileum (SIFileum), and Fed State Simulated Colonic Fluid (FeSSCoF). The compositions and preparation of these biorelevant dissolution media have been described previously.14,15)
Solubility of Gabapentin EnacarbilThe solubility of gabapentin enacarbil was measured in the dissolution media. One Regnite® tablets 300 mg was added to 20 mL of each biorelevant dissolution medium. The samples were shaken at approximately 200 shakes/min with a stroke length of 50 mm for 24 h at room temperature and subsequently filtered through a 0.45-µm polyvinylidene difluoride (PVDF) membrane (Whatman™ 13 mm GD/X, GE Healthcare UK Limited, Buckinghamshire, U.K.). The filtrate was immediately diluted with the mobile phase and analyzed by HPLC. Because they contain milk as a major component, FeSSGFmiddle samples cannot be filtered through 0.45-µm-sized pores, and filters with pore sizes of 2.7-µm (Whatman™ 25 mm Glass Microfiber GF/D) were used instead. FeSSGFmiddle filtrates were subsequently mixed well with the same volume of acetonitrile and the mixture was centrifuged at 16176×g for 5 min. The supernatant was analyzed by HPLC. All solubility measurements were conducted in duplicate.
In Vitro Dissolution TestingThree different devices were used to perform in vitro dissolution experiments: a paddle apparatus (USP Apparatus 2), the BioDis apparatus (USP Apparatus 3), and a flow-through cell apparatus (USP Apparatus 4).
Paddle ExperimentsA NTR-6400 type paddle dissolution apparatus (Toyama Sangyo Co., Ltd., Osaka, Japan) was used in this study. The dissolution media used were 300 mL of FaSSGF, or 500 mL of FeSSGFmiddle, FaSSIF-V2 or FeSSIF-V2 per vessel. Two different paddle revolutions at 50 or 100 rpm were applied. The temperature of the dissolution media in the vessels was maintained at 37±0.5°C throughout each test run. Samples (approximately 5 mL) were taken at 30, 60, 90, 120, 180, 240, 360, 480 and 1440 min, using a stainless cannula and plastic syringe. Samples were immediately filtered through a syringe filter of 0.45-µm PVDF membrane (Whatman™ 13 mm GD/X) into test tubes after discarding the first 2 mL of filtrate. Filtered solutions and the same volume of acetonitrile comprised samples for HPLC analysis. FeSSGFmiddle samples were pretreated using the same procedure as in the solubility assay. All dissolution experiments were conducted in triplicate.
BioDis ExperimentsA BIO-DIS III type reciprocating cylinder dissolution apparatus (Agilent Technologies, Inc., Santa Clara, CA, U.S.A.) was used in this study. The dissolution test set-up and dip rate are shown in Table 1. The temperature and volume of the dissolution media were 37±0.5°C and 220 mL, respectively. The detailed experimental procedures were performed according to previously reported methods.8–10) However, FeSSCoF, instead of FaSSCoF (Fasted State Simulated Colonic Fluid), was used as a colonic biorelevant medium for dissolution testing under both the fasted and fed states because the FaSSCoF medium was designed from human colonic contents after using bowel cleansing agent.16) We therefore concluded that it was reasonable to use FeSSCoF under the standard fasting condition in clinical trials. Further, the effect of share stress during gastric emptying was added to the dissolution test by increasing the dip rate to 30 dips/min for two minutes, given that several studies have shown that solid oral dosage forms can be exposed to mechanical pressures as high as 300 mbar during gastric emptying,11,17) and many studies have confirmed that the stress peaks in the antropyloric region.18–21) The tops and bottoms of the reciprocating cylinders were fitted with polypropylene mesh screens with mesh size of 405-µm (40 mesh) as a tablet holder. For the fasted state, samples were taken at 58, 60, 75, 90, 120, 240, 360 and 480 min. For the fed state, the samples were taken at 60, 120, 180, 238, 240, 270, 300, 420, 480, 540, 600 and 1440 min. Approximately 5 mL of samples were withdrawn using a stainless cannula and plastic syringe. All other conditions, including sample treatment, were the same as in the paddle experiment. Dissolution experiments were conducted in triplicate.
Table 1.
In Vitro Dissolution Set-Up of BioDis and Flow-through Cell Experiments
| Region | Medium | pH of medium | BioDis | Flow-through cell |
|---|
| Duration (min) | Dip rate (dips/min) | Duration (min) | Flow rate (mL/min) |
|---|
| Fasted state |
| Stomach | FaSSGF | 1.6 | 58 | 12 | 60 | 8 |
| 2 | 30 |
| Proximal gut | FaSSIF-V2 | 6.5 | 15 | 10 | 15 | 4 |
| Distal gut | FaSSIF-V2 | 6.8 | 15 | 10 | 15 | 4 |
| Midgut | FaSSIFmidgut | 6.8 | 30 | 10 | 30 | 4 |
| Distal ileum | SIFileum | 7.5 | 120 | 10 | 120 | 4 |
| Colon | FeSSCoF | 6.0 | 240 | 6 | 240 | 4 |
| Fed state |
| Stomach | FeSSGFmiddle | 5.0 | 238 | 15 | 240 | 8 |
| 2 | 30 |
| Proximal gut | FeSSIF-V2 | 5.8 | 30 | 10 | 30 | 4 |
| Midgut | FeSSIFmidgut | 6.5 | 30 | 10 | 30 | 4 |
| Distal ileum | SIFileum | 7.5 | 120 | 10 | 120 | 4 |
| Colon | FeSSCoF | 6.0 | 1020 | 6 | 120 | 4 |
A DFZ 720 type flow-through cell dissolution apparatus (Erweka GmbH, Heusenstamm, Germany) with large cells (22.6 mm diameter) was used in this study. A 5-mm-diameter glass bead was placed at the bottom of the cell. A total of 1.7 g of 1-mm-diameter glass beads were added to the 5-mm bead. In each cell, the Regnite® tablet 300 mg was placed on top of the 1-mm beads. A glass filter (Whatman™ 25 mm GF/F filter) was placed on top of each cell. The durations of the dissolution test were 8 and 9 h for simulating the fasted and fed states, respectively. The media were maintained at 37±0.5°C. The dissolution test set-up and flow rate are shown in Table 1. The detailed experimental procedures were performed according to previously reported methods.8–10) The dissolution tests were performed using an open-loop configuration with manual sample collection. For the fasted state, samples were taken at 20, 40, 60, 75, 90, 120, 165, 210, 240, 285, 320, 370, 420 and 480 min. For the fed state, samples were taken at 20, 40, 60, 80, 100, 120, 140, 160, 180, 210, 240, 255, 285, 330, 375, 405, 480 and 540 min. Approximately 5 mL of samples were withdrawn with a stainless cannula and plastic syringe at each time point. All other conditions were the same as those for the paddle experiment. Dissolution experiments were conducted in triplicate.
Analytical MethodsSamples obtained from the dissolution tests and solubility assessments were quantitatively analyzed to determine the concentration of gabapentin enacarbil by HPLC using the Alliance Separations Module 2695 with a type 2487 detector (Waters Corporation, Milford, MA, U.S.A.) at 35°C with a TSKgel ODS-100Z 5 µm column (4.6 mm i.d.×150 mm, Tosoh Corporation, Tokyo, Japan). The mobile phase was a mixture of acetonitrile, 0.02 M phosphate buffer (pH 2.5) and water (volume ratio 585 : 363 : 52). The flow rate was 1 mL/min and the injection volume was 10 µL. The detection wavelength was set at 210 nm. HPLC chromatograms were evaluated using Empower 3 (Waters Corporation).
Reference Pharmacokinetic DataBlood concentration–time profiles after oral administration of a gabapentin enacarbil IR capsule 350 mg in the fasted state were obtained from the literature.22) Similarly, plasma concentration-time profiles after oral dosing of a gabapentin enacarbil ER tablet 300 mg in the both fasted and fed states (U.S. Food and Drug Administration recommended high fat breakfast) were obtained from the literature.22)
Simulation of Plasma Concentration Profiles Using the Convolution MethodThe blood drug concentration–time profile of IR capsules in the fasted state was used to calculate the unit impulse response (UIR) function using Phoenix® WinNonlin® 6.3 (Certara, L.P., Princeton, NJ, U.S.A.). The PK profile of the IR capsule was described as a one-compartment model. The UIR function (A: 0.01433 L−1, α: 0.205821 h−1) was calculated from the PK profile of the IR capsule under the fasted state. To simulate the plasma concentration–time profiles of the gabapentin enacarbil ER tablet under both fasted and fed states, the dissolution profile at each prandial state was convoluted using the UIR function described above.
To conduct PK simulation of the ER tablet using paddle dissolution data, in vitro profiles in the intestinal media (FaSSIF-V2 or FeSSIF-V2) were superposed onto those in the stomach media (FaSSGF or FeSSGFmiddle) in consideration of the GI tract. The gastric emptying times in the superposed in vitro data were set to 1 h under the fasted state (FaSSGF to FaSSIF-V2) and 4 h under the fed state (FeSSGFmiddle to FeSSIF-V2), which matched the BioDis and flow-through cell settings.23,24) The paddle dissolution profiles in the stomach media were used up until set gastric emptying times of 1 h for the fasted state and 4 h for the fed state, respectively. The dissolution profiles in the intestinal media were corrected by multiplying the original values by undissolved ratio of the drug in the stomach media at the gastric emptying times. After the simulated gastric emptying time, the corrected dissolution profiles in the intestinal media from time 0 were superposed to the dissolved amount in FaSSGF or FeSSGFmiddle at the gastric emptying times. The superposed dissolution profiles were used to simulate the plasma profiles using convolution approach in the case of paddle apparatus, although the dissolution profiles from USP apparatus 3 and 4 were directly used for the simulation.
The PK parameters Tmax, Cmax and area under the plasma–drug concentration curve (AUC)0-inf were calculated for each simulated profile using non-compartmental analysis in Phoenix® WinNonlin® 6.3.
Evaluation of Prediction Accuracy of PK SimulationTo evaluate the prediction accuracy of each PK simulation, absolute percent prediction errors (%PE) for Cmax and AUC0-inf were calculated using the following Eq. 1:
 | (1) |
RESULTS AND DISCUSSION
Solubility of Gabapentin Enacarbil in Various Biorelevant MediaThe solubility of gabapentin enacarbil (Regnite® tablet) in biorelevant media using the shake flask method after 24 h of incubation at room temperature is shown in Fig. 1. Drug solubility in FaSSGF at pH 1.6 was approximately 0.7 mg/mL. This increased in SIFileum at pH 7.5 by about 13-fold (9.5 mg/mL), presumably because gabapentin enacarbil is a weakly acidic drug with a pKa of 5.025) and SIFileum has the highest pH among the media tested (Fig. 1a). The solubility of gabapentin enacarbil in FeSSIFmidgut (pH 6.5) was approximately 1.4-fold higher than that in FaSSIFmidgut (pH 6.8). It is likely that the higher concentrations of bile salts and lipids in the fed state media (FeSSGFmiddle, FeSSIF-V2 and FeSSIFmidgut) increased the solubility of the drug compared with the fasted state media.26,27) The solubility of gabapentin enacarbil in FeSSIF-V2 (pH 5.8) was higher than that in FeSSCoF (pH 6.0) despite the lower pH of FeSSIF-V2. This may be due to the higher concentration of bile acids contained in FeSSIF-V2.27)
We compared the solubility of gabapentin enacarbil in the fasted state stomach medium with that in the fed state medium. The solubility of gabapentin enacarbil in FeSSGFmiddle was approximately 5.5-fold higher than that in FaSSGF, presumably because the higher pH of FeSSGFmiddle improved solubility.
Dissolution TestsFigures 2a and 2c show the dissolution profiles of Regnite® tablets 300 mg in FaSSGF and FeSSGFmiddle at paddle revolution speeds of 50 rpm and 100 rpm, respectively. Figures 2b and 2d show the dissolution profiles in FaSSIF-V2 and FeSSIF-V2 at paddle revolution speeds of 50 rpm and 100 rpm, respectively. About 26 and 45% of the drug dissolved in 24 h in FaSSGF at 50 and 100 rpm, respectively. In FaSSIF-V2, about 62 and 82% of the drug dissolved in 24 h at 50 and 100 rpm, respectively. In contrast, only about 10 and 18% of the drug dissolved in FeSSGFmiddle at 50 and 100 rpm were in 24 h, respectively, although solubility of the drug in FeSSGFmiddle is higher than that in FaSSGF (Fig. 1). Milk protein in FeSSGFmiddle can potentially clabber when heated at 37°C, which might prevent penetration of the medium into the wax matrix and decrease the drug release from the wax matrix. In FeSSIF-V2, about 84 and 100% of the drug dissolved in 24 h at 50 and 100 rpm, respectively. The dissolution rate at 100 rpm was higher than that at 50 rpm for all media due to the stronger hydrodynamics at higher revolutions.
Despite the comparable solubility of gabapentin enacarbil in FaSSIF-V2 and FeSSIF-V2 (Fig. 1), dissolution in FeSSIF-V2 was faster than that in FaSSIF-V2. The glycerin fatty acid ester, used as a main component in ER formulations, may have undergone ester exchange with glyceryl monooleate contained in FeSSIF-V2.28) This may have caused a loosening of wax matrix structure, leading to faster dissolution.
Figures 2e and 2f show the dissolution profiles using BioDis and flow-through cell, respectively. In both BioDis and flow-through cell experiments, gabapentin enacarbil showed faster dissolution in simulated fasted state than fed state conditions. This may be explained by the fact that Regnite® tablet showed slow dissolution in the simulated gastric media under both prandial states (Figs. 2a and c) as well as a shorter residence time in the stomach under the fasted state.
The experimental conditions in the dissolution test, including the dip rate, flow rate, and duration for each dissolution medium, were slightly modified from those of previous reports,8–10) which were used to predict the in vivo performance of nifedipine ER tablets, two prototypes of colon-targeting tablets containing caffeine, and the diclofenac sodium oral modified-release pellet dosage form. In the present study, the hydrodynamic forces in these apparatuses had little effect on the dissolution rate of the drug up to a 1.7-fold dip rate and 2-fold flow rate (data not shown), while the paddle revolution rate strongly affected the dissolution profiles of the drug product. The dissolution did not reach 100% in 24 h in the simulated fed condition using BioDis. This suggests that the released but undissolved drug particles might pass through the mesh in the reciprocating cylinders, resulting in under-recovery of the drug, although according to the drug’s solubility (Fig. 1), the drug likely dissolves completely. In contrast, the dissolution was less than 40% at 8 h in both the fasted and fed conditions in the flow-through cell apparatus. This low fraction dissolved was probably due to the lower hydrodynamic forces in the flow-through cell system compared to the BioDis system, which prevented complete drug diffusion from the wax matrix.
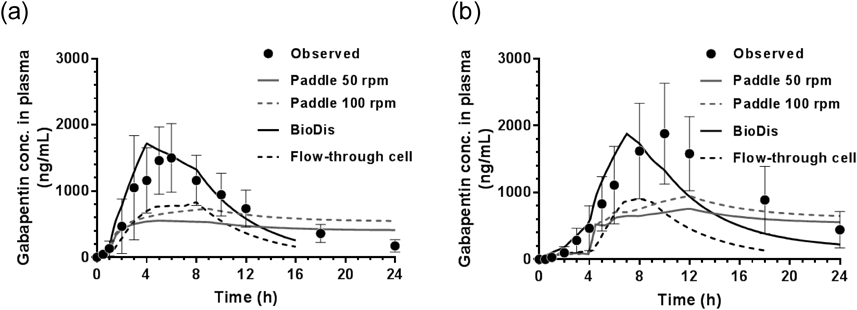
Simulations of Gabapentin Plasma Concentration ProfilesFigure 3 shows the simulated in vivo dissolution profiles obtained using in vitro paddle data at both 50 and 100 rpm for convolution analysis to predict oral PK profiles in humans. The paddle dissolution profiles in stomach media (Figs. 2a and c) were used up until set gastric emptying times of 1 h for the fasted state and 4 h for the fed state. The observed dissolution profiles in the intestinal media (Figs. 2b and d) were corrected by multiplying them by the undissolved ratio of the drug in the stomach media at the gastric emptying times. After the simulated gastric emptying time, these corrected dissolution profiles in the intestinal media were superposed to the dissolved amount in FaSSGF or FeSSGFmiddle at the gastric emptying time. For example, the paddle dissolution profile under 50 rpm in FaSSGF (Fig. 2a) was used until 1 h in which 2.1% of gabapentin dissolved. Then, the dissolution profile in the intestinal medium was corrected by multiplying the dissolution profile under 50 rpm in FaSSIF-V2 (Fig. 2b) from time 0 to 24 h by the undissolved ratio of 97.9%. Subsequently, the corrected dissolution profile under 50 rpm in FaSSIF-V2 was superposed on the drug dissolved in FaSSGF at 1 h (2.1%). Consequently, the simulated in vivo dissolution profile under the fasted state using paddle 50 rpm data (Fig. 3a) was obtained. Figures 2e (BioDis), 2f (flow-through cell), 3a (paddle 50 rpm), and 3b (paddle 100 rpm) were used as direct input functions and these dissolution profiles were convoluted using a sole UIR function to predict plasma concentration profiles of gabapentin after oral administration of a Regnite® tablets 300 mg.
Figure 4a and Table 2 show the predicted and observed PK profiles and parameters, respectively, under the fasted state. Predicted Cmax and AUC0-inf values with paddle revolutions of 50 rpm were underestimated by 67 and 27%, respectively. Similarly, these values with the flow-through cell were underestimated by 51 and 53%. In paddle revolutions of 100 rpm, there was little predictive error for AUC0-inf, whereas the %PE for Cmax value was 57%. Moreover, the predicted Tmax value was substantially delayed compared to the observed value and the predicted plasma profile was far from the observed data in Fig. 4a. In contrast, the predicted Cmax and AUC0-inf with BioDis were close to the observed values with their %PE values of within 15%, which is generally considered to be high prediction accuracy,29) for both PK parameters. The simulated Tmax was 0.83 h earlier than the observed mean value, suggesting that some subjects in clinical studies might exhibit slower gastric emptying of the gabapentin enacarbil ER tablet than the assumed 1 h in the in vitro dissolution test under the simulated fasted state. However, the simulated Tmax was within the standard deviation range of the observed Tmax value. These results suggest that the PK simulation using the BioDis data is the closest to the observed PK profile.
Table 2. Pharmacokinetic Parameters Estimated from Simulated Plasma Concentration Profiles
vs. Mean Observed Values with Standard Deviations (in Parentheses) after Oral Administration of a Regnite
® Tablets 300 mg under the Fasted State
| Tmax (h) | Cmax (µg/mL) | AUC0-inf (µg·h/mL) | %PE |
|---|
| Cmax | AUC0-inf |
|---|
| Observed | 4.85 (1.41) | 1.69 (0.506) | 18.0 (6.26) | | |
| Predicted (paddle 50 rpm) | 5.03 | 0.55 | 13.1 | 67 | 27 |
| Predicted (paddle 100 rpm) | 9.05 | 0.73 | 17.1 | 57 | 5 |
| Predicted (BioDis) | 4.02 | 1.72 | 15.7 | 2 | 13 |
| Predicted (flow-through cell) | 7.96 | 0.83 | 8.4 | 51 | 53 |
Figure 4b and Table 3 show the predicted and observed PK profiles and parameters, respectively, under the fed state. Predicted Cmax values with paddle revolutions of 50 and 100 rpm were underestimated by 67 and 58%, respectively. Similarly, the %PE of AUC0-inf values were 36 and 24%, respectively. The %PE values with flow-through cell were 60% for Cmax and 72% for AUC0-inf. In contrast, the %PE of Cmax value with BioDis was 17%, which was the closest to the observed data among the dissolution methods studied and the %PE of AUC0-inf was 33%. The simulated value of Tmax under the fed state was earlier than the observed mean value, similar to that for the fasted state. Our findings suggest that the predicted apparent absorption duration of gabapentin is shorter than the observed durations. The %PE values for both Cmax and AUC0-inf were higher than 15%, suggesting inaccurate prediction accuracy under the fed state. Although the simulated fed state dissolution media have been used in this study, many factors (e.g. hydrolysis of the prodrug, GI transit, hydrodynamics, shear forces and physical pressures, etc.) under the fed state need to be optimized for more precise prediction. Another possible reason for the inaccurate prediction under the fed state would be that the UIR function from the PK data under the fasted state was used in the simulation for both prandial state.
Table 3. Pharmacokinetic Parameters Estimated from Simulated Plasma Concentration Profiles
vs. Mean Observed Values with Standard Deviations (in Parentheses) after Oral Administration of a Regnite
® Tablets 300 mg under the Fed State
| Tmax (h) | Cmax (µg/mL) | AUC0-inf (µg·h/mL) | %PE |
|---|
| Cmax | AUC0-inf |
|---|
| Observed | 9.83 (2.76) | 2.26 (0.552) | 27.4 (6.29) | | |
| Predicted (paddle 50 rpm) | 11.82 | 0.76 | 17.7 | 67 | 36 |
| Predicted (paddle 100 rpm) | 11.82 | 0.94 | 20.8 | 58 | 24 |
| Predicted (BioDis) | 6.99 | 1.88 | 18.5 | 17 | 33 |
| Predicted (flow-through cell) | 7.96 | 0.91 | 7.8 | 60 | 72 |
Based on the above results, the in vivo performance of gabapentin enacarbil ER tablets was best predicted by BioDis among the in vitro dissolution apparatuses under the current experimental conditions. In the paddle method, because a fixed volume of a single dissolution medium is often used, it was difficult to simulate the transit of an ER dosage form through the GI tract. In contrast, BioDis and flow-through cell methods could simulate the GI tract environment by changing the test media smoothly. In terms of hydrodynamics, a previous study suggested that the current flow-through cell conditions might be too low for both the fasted and fed conditions.9) In contrast, the hydrodynamic force used in BioDis seemed to closer to in vivo values than that used in the flow-through cell. Regnite® tablets 300 mg is a wax matrix-type ER formulation and drug release occurs following both erosion of the wax matrix component and diffusion of the drug.30,31) The mechanism of the erosion of the wax matrix component is presumed to be as follows: in vitro dissolution media or in vivo GI fluids penetrate into the wax matrix, leading to disintegration of the matrix from the surface of the tablet and subsequent dissolution and release of the drug from the resultant solid particles. In contrast, the mechanism of diffusion is presumed to be as follows: dissolution media or GI tract fluids penetrate into the wax matrix, dissolving the drug, leading to its diffusion through the wax matrix. Our findings suggest that the BioDis method may accurately simulate the erosion and diffusion of the ER formulation in the GI tract in vivo.
While various in vitro dissolution conditions can be employed by adjusting the dip and the flow rates using BioDis and flow-through cell methods, respectively, there are limitations to mimicking the in vivo dissolution of ER formulations. In addition to simulating the composition of GI fluids, drug release and dissolution in in vivo can also be influenced by transit time, hydrodynamics, shear force and physical pressures in the GI tract. It is challenging to simulate shear forces and pressures using the compendial dissolution equipment like BioDis and flow-through cell. In particular, additional investigation is needed for solid dosage forms that are sensitive to shear force and pressures. The in vivo performance of dosage forms might be better predicted using more complicated dissolution systems, such as the dynamic gastric model32) and stress test device.11,12)
CONCLUSION
Biorelevant dissolution using BioDis coupled with a convolution approach successfully predicted the PK profile of gabapentin enacarbil after oral administration of an ER tablet under the fasted state. Among the dissolution methods studied, the predicted PK profile using the BioDis data was closest to the observed one under the fed state, however, that prediction was not adequate accuracy with large prediction errors. This approach might be useful for predicting oral PK profiles of other drugs formulated as wax matrix-type ER tablets under the fasted state.
Conflict of Interest
Satomi Yamaguchi Ikeuchi, Atsushi Kambayashi and Hiroyuki Kojima are employees of Astellas Pharma Inc. The other authors declare no conflict of interest.
REFERENCES
- 1) Nicolaides E, Symillides M, Dressman JB, Reppas C. Biorelevant dissolution testing to predict the plasma profile of lipophilic drugs after oral administration. Pharm. Res., 18, 380–388 (2001).
- 2) Shono Y, Jantratid E, Janssen N, Kesisoglou F, Mao Y, Vertzoni M, Reppas C, Dressman JB. Prediction of food effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur. J. Pharm. Biopharm., 73, 107–114 (2009).
- 3) Wagner C, Jantratid E, Kesisoglou F, Vertzoni M, Reppas C, Dressman JB. Predicting the oral absorption of a poorly soluble, poorly permeable weak base using biorelevant dissolution and transfer model tests coupled with a physiologically based pharmacokinetic model. Eur. J. Pharm. Biopharm., 82, 127–138 (2012).
- 4) Tsume Y, Mudie DM, Langguth P, Amidon GE, Amidon GL. The Biopharmaceutics Classification System: subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci., 57, 152–163 (2014).
- 5) Okumu A, DiMaso M, Lobenberg R. Computer simulations using GastroPlus to justify a biowaiver for etoricoxib solid oral drug products. Eur. J. Pharm. Biopharm., 72, 91–98 (2009).
- 6) Klein S, Rudolph MW, Skalsky B, Petereit HU, Dressman JB. Use of the BioDis to generate a physiologically relevant IVIVC. J. Control. Release, 130, 216–219 (2008).
- 7) Fotaki N, Aivaliotis A, Butler J, Dressman J, Fischbach M, Hempenstall J, Klein S, Reppas C. A comparative study of different release apparatus in generating in vitro–in vivo correlations for extended release formulations. Eur. J. Pharm. Biopharm., 73, 115–120 (2009).
- 8) Andreas CJ, Chen YC, Markopoulos C, Reppas C, Dressman J. In vitro biorelevant models for evaluating modified release mesalamine products to forecast the effect of formulation and meal intake on drug release. Eur. J. Pharm. Biopharm., 97 (Pt A), 39–50 (2015).
- 9) Andreas CJ, Tomaszewska I, Muenster U, van der Mey D, Mueck W, Dressman JB. Can dosage form-dependent food effects be predicted using biorelevant dissolution tests? Case example extended release nifedipine. Eur. J. Pharm. Biopharm., 105, 193–202 (2016).
- 10) Andreas CJ, Pepin X, Markopoulos C, Vertzoni M, Reppas C, Dressman JB. Mechanistic investigation of the negative food effect of modified release zolpidem. Eur. J. Pharm. Sci., 102, 284–298 (2017).
- 11) Garbacz G, Wedemeyer RS, Nagel S, Giessmann T, Monnikes H, Wilson CG, Siegmund W, Weitschies W. Irregular absorption profiles observed from diclofenac extended release tablets can be predicted using a dissolution test apparatus that mimics in vivo physical stresses. Eur. J. Pharm. Biopharm., 70, 421–428 (2008).
- 12) Garbacz G, Weitschies W. Investigation of dissolution behavior of diclofenac sodium extended release formulations under standard and biorelevant test conditions. Drug Dev. Ind. Pharm., 36, 518–530 (2010).
- 13) Lal R, Sukbuntherng J, Luo W, Tovera J, Lassauzet ML, Cundy KC. Population pharmacokinetics and pharmacodynamics of gabapentin after administration of gabapentin enacarbil. J. Clin. Pharmacol., 53, 29–40 (2013).
- 14) Vertzoni M, Dressman J, Butler J, Hempenstall J, Reppas C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur. J. Pharm. Biopharm., 60, 413–417 (2005).
- 15) Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm. Res., 25, 1663–1676 (2008).
- 16) Diakidou A, Vertzoni M, Goumas K, Soderlind E, Abrahamsson B, Dressman J, Reppas C. Characterization of the contents of ascending colon to which drugs are exposed after oral administration to healthy adults. Pharm. Res., 26, 2141–2151 (2009).
- 17) Cassilly D, Kantor S, Knight LC, Maurer AH, Fisher RS, Semler J, Parkman HP. Gastric emptying of a non-digestible solid: assessment with simultaneous SmartPill pH and pressure capsule, antroduodenal manometry, gastric emptying scintigraphy. Neurogastroenterol. Motil., 20, 311–319 (2008).
- 18) Kozu H, Kobayashi I, Nakajima M, Uemura K, Sato S, Ichikawa S. Analysis of flow phenomena in gastric contents induced by human gastric peristalsis using CFD. Food Biophys., 5, 330–336 (2010).
- 19) Boulby P, Moore R, Gowland P, Spiller RC. Fat delays emptying but increases forward and backward antral flow as assessed by flow-sensitive magnetic resonance imaging. Neurogastroenterol. Motil., 11, 27–36 (1999).
- 20) Pal A, Brasseur JG, Abrahamsson B. A stomach road or “Magenstrasse” for gastric emptying. J. Biomech., 40, 1202–1210 (2007).
- 21) Ferrua MJ, Kong FB, Singh RP. Computational modeling of gastric digestion and the role of food material properties. Trends Food Sci. Technol., 22, 480–491 (2011).
- 22) Cundy KC, Sastry S, Luo W, Zou J, Moors TL, Canafax DM. Clinical pharmacokinetics of XP13512, a novel transported prodrug of gabapentin. J. Clin. Pharmacol., 48, 1378–1388 (2008).
- 23) Klein S, Stein J, Dressman J. Site-specific delivery of anti-inflammatory drugs in the gastrointestinal tract: an in-vitro release model. J. Pharm. Pharmacol., 57, 709–719 (2005).
- 24) Klein S. Predicting food effects on drug release from extended-release oral dosage forms containing a narrow therapeutic index drug. Dissolut. Technol., 16, 28–40 (2009).
- 25) Cundy KC, Branch R, Chernov-Rogan T, Dias T, Estrada T, Hold K, Koller K, Liu X, Mann A, Panuwat M, Raillard SP, Upadhyay S, Wu QQ, Xiang JN, Yan H, Zerangue N, Zhou CX, Barrett RW, Gallop MA. XP13512 [(+/−)-1-([(alpha-isobutanoyloxyethoxy)carbonyl]aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther., 311, 315–323 (2004).
- 26) Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm. Res., 13, 163–167 (1996).
- 27) Markopoulos C, Andreas CJ, Vertzoni M, Dressman J, Reppas C. In-vitro simulation of luminal conditions for evaluation of performance of oral drug products: Choosing the appropriate test media. Eur. J. Pharm. Biopharm., 93, 173–182 (2015).
- 28) Otera J. Transesterification. Chem. Rev., 93, 1449–1470 (1993).
- 29) FDA. Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of in Vitro/in Vivo Correlations (1997).
- 30) Agata Y, Iwao Y, Shiino K, Miyagishima A, Itai S. A theoretical approach to evaluate the release rate of acetaminophen from erosive wax matrix dosage forms. Int. J. Pharm., 414, 63–68 (2011).
- 31) Nitanai Y, Agata Y, Iwao Y, Itai S. A novel mathematical model considering change of diffusion coefficient for predicting dissolution behavior of acetaminophen from wax matrix dosage form. Int. J. Pharm., 428, 82–90 (2012).
- 32) McAllister M. Dynamic dissolution: a step closer to predictive dissolution testing? Mol. Pharm., 7, 1374–1387 (2010).