Review
Systems Pathology of Neuropathic Pain and Fibromyalgia
2019 Volume 42 Issue 11 Pages 1773-1782
Details
2019 Volume 42 Issue 11 Pages 1773-1782
Currently, only a few medicines have been approved for use in the clinical treatment of chronic pain, but they are not fully satisfying due to their side effects. From the view that radical treatment, rather than simply treating symptoms, is more important in addressing life-long chronic pain, we have been investigating translational research for a mechanism-based medicine to treat pain. Through the characterization of various types of peripheral and central neuropathic pain in mice, we discovered that lysophosphatidic acid (LPA) plays roles in definitive mechanisms of the development and maintenance of neuropathic pain. We found LPA1 receptor- and LPA3 receptor-mediated amplification of LPA production could be a key mechanism underlying the initiation and maintenance of this pain. We have developed stress-induced fibromyalgia models, and have revealed that LPA1 receptor-signaling also plays key roles in the mechanism. Throughout these studies, we found that LPA plays a key role in pain memory, and that LPA1 receptor- and LPA3 receptor-antagonists could reverse the established pain, and thereby cure the disease source of pain.
Pain has been broadly categorized into acute pain and chronic pain (Fig. 1). Acute pain includes nociceptive (or physiological) and inflammatory pain, both of which are suppressed by opioid analgesics and anti-inflammatory drugs. Inflammatory pain following tissue damage is reversible when the cause is removed (i.e., the tissue heals). In contrast, chronic pain is in general refractory, and lasts for long periods, even after recovery from the initial cause. One such representative chronic pain is neuropathic pain, which is caused by neuronal damage within the pain pathway from primary afferent nerves to higher brain areas via the spinal cord. Pain, in most cases, is observed in the area closely related to the pain pathway. Another newly proposed category of chronic and refractory pain is “centralized pain,” here represented by the generalized pain disease of fibromyalgia. The cause of centralized pain remains elusive, but it has been postulated to include a history of intense physical and/or emotional stress. As such, centralized pain is now discussed in relation to the brain’s memory of pain. This review provides an overview of studies we have been working on regarding chronic pain and its mechanisms.
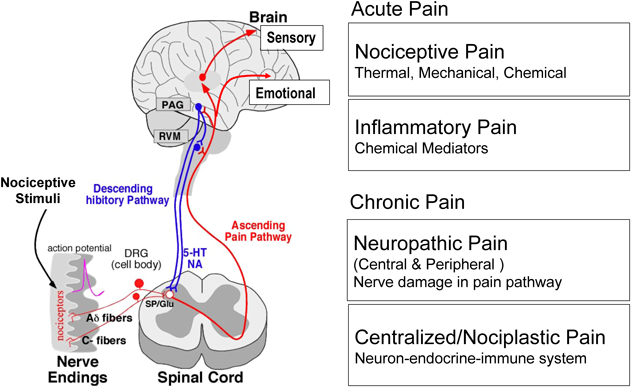
Details are described in the text. (Color figure can be accessed in the online version.)
Pain has a role in alarming us, to let us know of tissue damage so that in the future we attempt to avoid or escape such painful danger signals. Most instant physiology in relation to this escape behavior seems to be a spinal reflex. Acute pain perception is mediated through sensory unmyelinated C- and thinly myelinated Aδ-fibers, which innervate superficial spinal dorsal horn neurons (Rexed Lamina I and II layers), which cross the spinal cord for spinothalamic pain transmission, and at the same time connect to ventral neurons for the nociceptive reflex. In general, innocuous or tactile perception is known to activate sensory Aβ-fibers, which mainly innervate neurons at the cuneate and gracile nucleus in the medulla oblongata, while a small portion of Aβ-fibers innervate deeper spinal neurons (Lamina III, IV and deeper layers). Therefore, the roles of sensory Aβ-fibers in the spinal reflex seem to be less important compared to the roles of C- and Aδ-fibers; therefore, reflex or paw withdrawal behaviors have been used to assess pain/nociceptive responses in experimental animals. Nociceptive responses to thermal, mechanical and chemical stimuli have been used for many years in rodent experiments, such as the Hargreaves’ or thermal paw withdrawal test,1) the von Frey filament or paw pressure test,2,3) and the formalin test,4) respectively.
2.2. Pharmacologically Defined Algogenics-Induced Nociception TestAs we experience thermal, mechanical and chemical stimuli, the nociception tests using these stimuli are advantageous to properly evaluate pharmacological actions against acute pain. However, it is difficult to examine the nociceptive or antinociceptive responses of new compounds and their mechanisms by the above-mentioned nociception tests. For this purpose, we devised an algogenics-induced paw flexion (APF) test in mice, in which the mouse is put in a cloth hammock bag with holes for exposure of its 4 legs.5,6) A very thin plastic tube is inserted into the paw of one hind limb, and very small amounts of pharmacologically defined chemicals are infused into the paw. We can evaluate nociceptive or anti-nociceptive responses by combining the test chemical with well-defined pain-producing compounds. One of several advantages of the APF test is its high sensitivity, which may be explained by the view that pain-suppressive tactile information is cancelled by hanging the hammock. Another advantage is the accessibility to studying in vivo signal transduction by the use of intrathecal (i.t.) pretreatment with antisense oligos or small interfering RNA (siRNA) for knockdown of specific protein expression in dorsal root ganglion (DRG) and peripheral nociceptor endings. Further, we evaluated sensitivities to neonatal capsaicin pretreatment7–9) or intrathecal substance P or glutamate receptor antagonism. Thus, we proposed a new categorization of pain fibers into three types: type I C-fibers, which are sensitive to neonatal capsaicin and spinal NK1 receptor antagonist (i.t.); type II C-fibers, which are sensitive to neonatal capsaicin-sensitive and N-methyl-D-aspartate (NMDA) receptor antagonist (i.t.); and Aδ-fibers, which are not sensitive to neonatal capsaicin, but sensitive to the NMDA receptor antagonist (i.t.).10–12) Algogenics that activate type I C-fibers are substance P (NK1 receptor), bradykinin (B2 receptor) and histamine (H1 receptor) through Gq/11-coupled receptors. A type II C-fiber activator is ATP (P2X3 receptor). And Aδ-fiber activators include the prostaglandin (PG)I2 receptor agonist, ONO-54918-07, or the tuberoinfundibular peptide of 39 residues (TIP39), which is an endogenous PTH2 receptor ligand,13) respectively.
2.3. Sensory Fiber-Specific Nociception TestTo evaluate Aβ-specific responses, an assessment not included in the APF test, we have adopted a method using a Neurometer, which is presumed to separately stimulate C-, Aδ- and Aβ-fibers by use of differential frequencies of electrical stimulation at 5250 and 2000 Hz, respectively.14–16) In the electrical stimulation-induced paw flexor test (EPF), the mouse is also kept in a hammock-type cloth-container, while the foot is stimulated by a neurometer through a set of electrodes, one at the sole, the other on the instep.17) We evaluate the threshold of currents that cause paw withdrawal behavior. Although the nociceptive threshold is very reproducible, the way to calm the mouse in a cloth container requires an experimenter’s expertise; therefore, we improved this test, creating an electrical paw withdrawal (EPW) test in which the mouse is held in the experimenter’s hand, and its foot given electrical stimulation, followed by evaluation of the threshold to exhibit paw withdrawal.15) The several advantages to EPW testing is as follows: 1) no adaptation time is necessary, 2) C, Aδ and Aβ responses are separately evaluated, and 3) the nociceptive threshold is reproducible.
Neuropathic pain in experimental animal models is characterized by allodynia or hyperalgesia, where normally innocuous or mildly noxious stimuli induce a nociceptive (pain) behavioral response. In peripheral nerve injury models, partial ligation of the sciatic nerve (pSNL) is often used to induce neuropathic pain behavior, and the presence of neuropathic pain is mainly assessed by hyperalgesia to thermal and mechanical stimuli, and allodynia to cold and tactile stimuli.18,19) When APF, EPF or EPW tests were used, we found several unique findings in the sensory fiber-specific responses. The type-1 C-fiber-mediated responses to substance P, bradykinin and histamine were all abolished, while type-2 C-fiber-mediated responses to ATP were not affected. On the other hand, Aδ-mediated responses to TIP39 or the PGI2 agonist, ONO-54918-07, were 100- to 1000-fold enhanced.20,13) The loss of function or so-called “hypoesthesia” through C-fibers was also observed in the EPF or EPW test, while hyperalgesia was consistently observed in the responses mediated through Aδ and Aβ-fibers.6)
3.2. Demyelination in Mice with Peripheral InjuryIn the studies involving EPF or EPW testing, pSNL-induced neuropathic hyperalgesia and allodynia were observed with the stimulation of Aδ- and Aβ-fibers, respectively, while unique hypoesthesia occurred in the case of C-fiber stimulation. Accordingly, we speculated that nerve injury-induced painful alteration may be related to the demyelination of A-fibers. Furthermore, it is known that signals from Schwann cells suppress neurite growth, while demyelination is reported to cause sprouting, which may facilitate the cross-talk between Aβ-fibers and C- or Aδ-fibers, thereby forming abnormal sensory innervation of sprouted Aβ-fibers to the spinal neurons, which had been innervated to C- or Aδ-fibers. We were successful in demonstrating physical cross-talk between A-fibers and C-fibers in the Remak bundle16) and abnormal synapses, as measured by the phosphorylation of ERK1/2 in spinal dorsal horn neurons after pSNL.15) The synaptic reorganization seen in the latter case may be facilitated by the loss of C-fiber function (hypoesthesia), which may be explained by the substance P fiber retraction at the central ends of C-fibers19) or by the down-regulation of voltage-dependent Na channels, such as Nav1.8.21) The Nav1.8 downregulation is explained by two different molecular mechanisms: through the loss of nerve growth factor (NGF) supply,22) and by epigenetic silencing of Nav1.8 in the DRG.23)
3.3. LPA Involvement Underlying Abnormal Pain BehaviorsCircumstantial evidence suggests that LPA1 receptor signaling causes the retraction of neuronal growth cones24) and morphological changes in Schwann cells.25) These changes are presumed to play roles in C-fiber retraction26) and A-fiber demyelination with subsequent sprouting.27) The experimental observations supporting this speculation are as follows: 1) pSNL causes LPA1 receptor-mediated demyelination of dorsal root fibers through RhoA and Rho-associated protein kinase (ROCK) mechanisms28); 2) the addition of LPA to an ex vivo culture of peripheral nerve fibers causes significant demyelination29); 3) the addition of LPA to cultured S16 Schwann cells causes gene silencing of myelin protein through a ROCK-mediated pathway30); 4) the addition of LPA causes demyelination and sprouting of myelinated fibers in a co-culture of separately isolated DRG neurons and Schwann cells6); 5) injury or intrathecally administered LPA induces the degradation of myelin-associated glycoprotein (MAG) by LPA1 receptor-mediated calpain activation; and 6) pSNL causes an Aβ-fiber stimulation-induced rapid phosphorylation of ERK1/2 in neurons at the substantia gelatinosa of the dorsal horn, where C- or Aδ-fibers innervate, through LPA1 receptor-mediated mechanisms.31) All these mechanisms may properly explain the schematic and hypothetical models of mechanisms underlying neuropathic pain behaviors (Fig. 2).
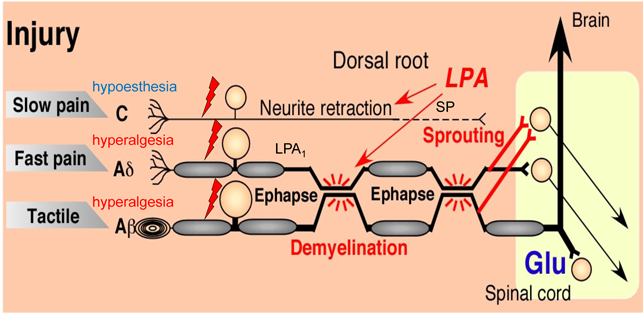
Details are described in the text. Briefly, functional changes after partial sciatic nerve ligation (pSNL) included hypoesthesia via C-fibers and hyperalgesia via Aδ and Aβ fibers. Morphological changes included the retraction of substance P-containing C-fibers and the demyelination of A-fibers. There is functional evidence (PMID) showing that sprouts from demyelinated Aβ-fibers may innervate spinal neurons, which had also been innervated by C- or Aδ-fibers. (Color figure can be accessed in the online version.)
Extracellular LPA is easily degraded by membrane-bound lipid phosphate phosphatases. Several studies have reported that the half-life of 32P-LPA is 5 min or less in both ex vivo and in vivo preparations.32,33) However, a single intrathecal (i.t.) injection of LPA causes thermal and mechanical hyperalgesia for a week.28,34) Therefore, we attempted to solve this key question of why LPA (i.t.) causes such long-lasting actions. From several attempts, we found that LPA (i.t.) causes LPA production through microglia activation.35–37) LPA-induced LPA production was also supported by in vitro experiments using spinal cord slices. These findings led to several discoveries, allowing for the development of a new concept for the feed-forward mechanism of LPA production following peripheral nerve injury (Fig. 3).
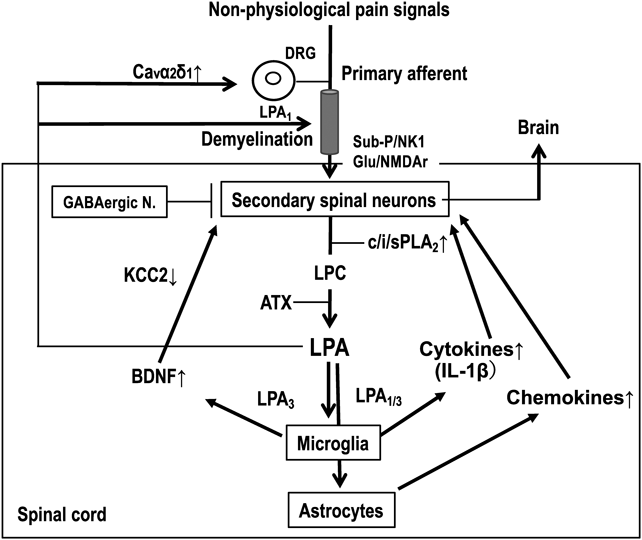
Details are described in the text.
The first step in our hypothesis38) is related to the view that peripheral nerve injury causes intense non-physiological pain signals, or simultaneously transmits the information from different types of sensory fibers to the spinal dorsal horn neurons, in contrast to the case with tissue inflammation, which produces chemical mediators that subsequently activate specific sensory fibers (e.g. C-fibers). As a result, secondary spinal neurons will be abnormally activated, and produce abundant lysophosphatidylcholine (LPC) by an action of not only cytosolic and Ca-dependent phospholipase A2 (cPLA2), but also by Ca-insensitive phospholipase A2 (iPLA2), which maintains the production of LPC even after the depletion of intracellular Ca levels.39) As LPC is an amphiphilic compound, locally concentrated LPC may form micelles, which in turn are excreted into the extracellular space. As significant amounts of autotaxin (ATX), which has lysophospholipase D activity, exist in cerebrospinal fluid, LPC is immediately converted to LPA. Thus, the possible involvement of LPC export through transporters cannot be excluded, although no evidence of this is currently available.
3.4.2. LPA1-Signaling Distinguishes Neuropathic Pain from Inflammatory PainIn our preliminary studies, nerve injury has significantly increased the LPA levels in the spinal dorsal horn, while Complete Freund’s Adjuvant (CFA)-treated inflammation does not. Such a possible difference between nerve injury and tissue inflammation in terms of LPA production was supported in a behavioral study, in which the genetic deficiency of LPA1 receptor completely abolished the mechanical hyperalgesia (neuropathic pain) at day 3 or later after pSNL-treatment,28) while there was a very limited blockade of mechanical hyperalgesia induced by CFA-treatment (a Rheumatoid Arthritis-type inflammatory pain model), as shown in Fig. 4. When the pain thresholds at days 1, 3, 5, 7 and 9 after CFA were calculated, the hyperalgesia was inhibited by only 22% in LPA1 receptor-KO mice. Of most interest, the inhibition of hyperalgesia at day 1 after pSNL in LPA1 receptor-KO mice was very little, suggesting the initial hyperalgesia after pSNL is attributed to damage-induced inflammation. Very weak suppression of inflammatory pain in LPA1 receptor-KO mice was supported by the previous report using the inflammatory orofacial pain model and LPA1 receptor antagonist AM095.40)
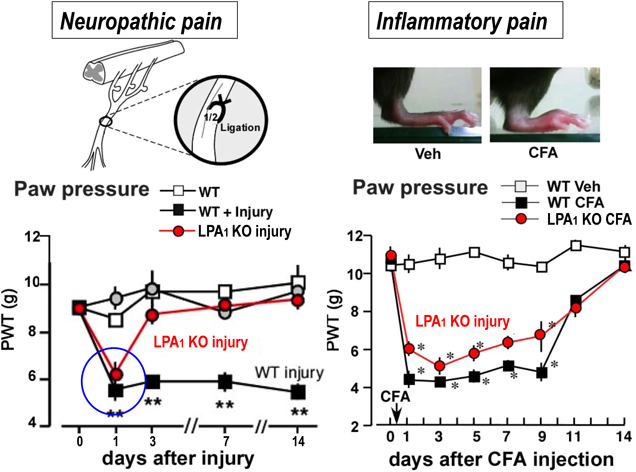
The pSNL-induced neuropathic mechanical hyperalgesia lasted for more than 14 d. This long-lasting neuropathic pain was completely abolished in LPA1 receptor-KO mice, except for day 1. The inflammatory pain caused by intraplantar injection of Complete Freund’s Adjuvant (CFA) lasted for 9 d. However, a large portion of this hyperalgesia remained, although some portion was inhibited in LPA1 receptor-KO mice. (Color figure can be accessed in the online version.)
The following step is related to the involvement of microglia in LPA production.37) LPA activates microglia and produces interleukin-1β (IL-1β),41) which in turn stimulates both cPLA2 and iPLA2,42) leading to further LPA production (feed-forward LPA production). Some aliquots of LPA may go back to the dorsal root fibers and DRG, where LPA causes LPA1 receptor-mediated demyelination (allodynia) and upregulation of Cavα2δ1 (hyperalgesia due to enhanced spinal transmission), respectively.43) Enhanced pain mechanisms through LPA-actions on primary afferents are presumed to reinforce the feed-forward LPA production in the spinal cord, as well as abnormal pain transmission. In a culture experiment, LPA acted on microglia and released ATP, which in turn activated the P2X4 receptor and increased BDNF gene expression.44) BDNF is known to decrease neuronal plasma membrane KCC2 levels and increase cytosolic Cl− ion levels, resulting in a conversion of GABAA receptor function from an inhibitory to an excitatory one.45,46) As microglia inhibitors block neuropathic pain,47) all these mechanisms may play roles in the development of neuropathic pain.
3.5. LPA Receptor-Mediated Maintenance of Neuropathic Pain through Glial ActivationA significant increase in LPA production at the dorsal horn was also observed at 2 and 3 weeks after pSNL, suggesting that LPA1 and/or LPA3 receptor-mediated feed-forward LPA production and “Pain Memory” mechanisms are maintained. When the LPA1/3 receptor antagonist Ki14625 was given i.t. from day 7 to day 13 after pSNL, there was a time-dependent gradual reverse of basal pain threshold to the normal level in the thermal nociception test (Fig. 5), as reported.48) In addition, astrocyte activation with chemokine CXCL1 overexpression was detected at day 14 after the pSNL. As these changes were blocked by Ki14625, it is safe to say that LPA-mediated astrocyte activation may be related to pain mechanisms at the later stage (or to the maintenance of neuropathic pain). This view was further supported by the finding that the hyperalgesia was significantly inhibited by pretreatment with the astrocyte-specific toxin L-α-aminoazipate (L-aa), which selectively decreases the population of astrocytes, but not microglia, according to flow cytometry analysis. However, LPA production was blocked by pretreatment with Mac1-Saporin, a microglia toxin, but not by L-aa. Therefore, at the later stage of neuropathic pain, LPA production seems to be maintained by microglia actions, while the pain production may be related to LPA receptor-mediated astrocyte activation, consistent with the previous report describing neuronal-glial interaction underlying neuropathic pain.49) When primary cultured astrocytes were treated by LPA, various chemokines were upregulated. In addition, the intrathecal administration of LPA-primed astrocytes caused significant hyperalgesia in an LPA1 receptor-mediated mechanism.48)
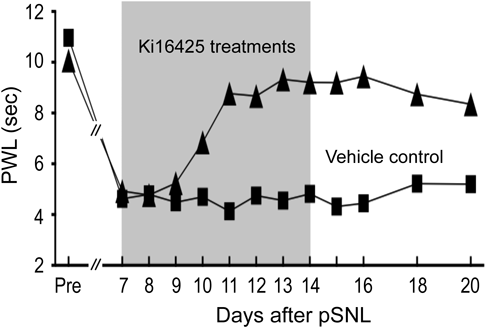
In the pSNL-induced neuropathic pain model, established thermal hyperalgesia was completely cured by repeated treatments with Ki16425, an LPA1 and LPA3 receptor antagonist (30 mg/kg i.p., twice daily). Note that the blockade of neuroparthic pain still remained for another week after the cessation of Ki16425 treatments.
There are many reports that chemotherapeutic agents represented by paclitaxel cause peripheral neuropathic pain.50–52) This is a unique type of chronic pain, since this pain would potentially be treated in a prophylactic way. The abnormal microtubule assembly of paclitaxel is considered one of the molecular mechanisms underlying its toxicity. It is interesting to consider that there is a relationship between peripheral neuropathic pain and longer axons of sensory fibers. In the case of paclitaxel therapy, mitochondrial damage may be secondary to the abnormal tubulin assembly-related disturbance of axonal flow. In the analogy of nerve injury-induced neuropathic pain mechanisms, we tested the effects of LPA1 and LPA3 receptor-KO mice on paclitaxel-induced peripheral neuropathic pain. Hyperalgesia was observed 1 d after the initial paclitaxel treatment (4 mg/kg i.p.), and this lasted for more than 2 weeks by treatments given 4-times, every other day. The hyperalgesia disappeared in LPA1- and LPA3 receptor-KO mice throughout the experiments. As expected, repeated pretreatment with Ki16425 twice daily (13-times in total) abolished the neuropathic pain. LPA production was also significantly increased at 24h after the first paclitaxel administration, while cPLA2 and iPLA2 activation were observed with a peak time at 12h for both enzymes.51) When we assessed the morphology of paclitaxel-treated sensory fibers by transmission electron microscopic (TEM) analysis, a swollen endometrium and debruised myelin sheath lamellae indicated degenerated myelin fibers, and atrophy of the axon were observed in dorsal root A-fibers, but not in spinal nerve or sciatic nerve fibers, as shown in Fig. 6. Toluidine blue analysis showed the quantitative increase in demyelination, and Western blot analysis showed a significant decrease in myelin protein MAG levels at the dorsal root. Furthermore, the demyelination was significantly attenuated in LPA1-, LPA3 receptor-KO and heterozygous ATX-KO mice.
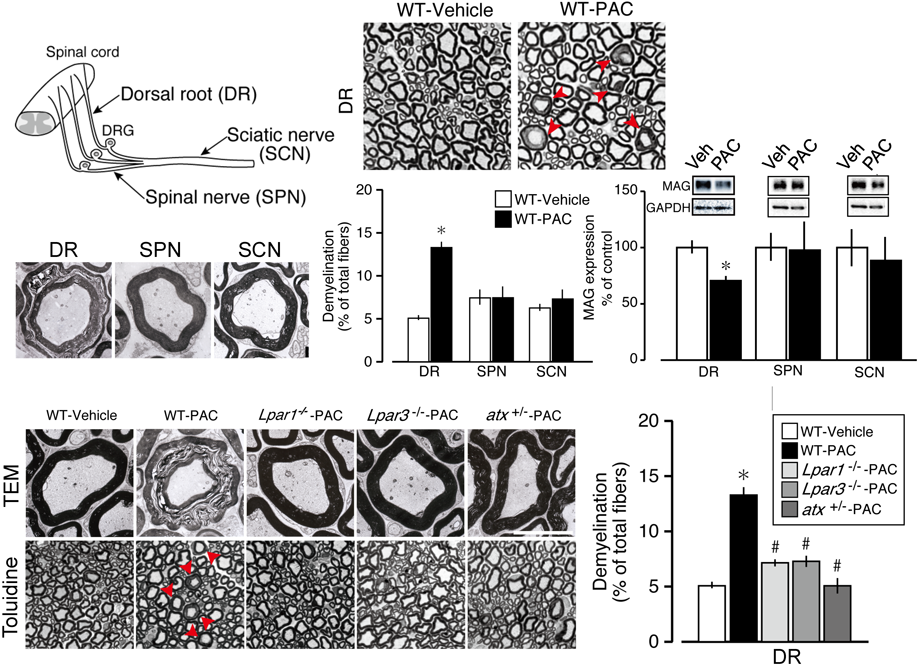
Mice with repeated paclitaxel treatments (4 times each, on days 0, 2, 4, and 6) showed long-lasting hyperalgesia, which was abolished in LPA1 receptor- and LPA3 receptor-KO mice (PMID). In the same model, dorsal root (DR)-specific demyelination and downregulation of the myelin protein MAG were observed. Demyelination was significantly attenuated in LPA1 receptor- and LPA3 receptor-KO mice, as well as in heterozygous ATX-KO mice. Details are described in the text. All data are unpublished (by Ueda, H, Uchida, H, and Yano, R). (Color figure can be accessed in the online version.)
Central post stroke pain (CPSP) is a representative central neuropathic pain. We successfully developed the CPSP model in mice using photochemically induced thrombosis (PIT) in combination with tissue plasminogen-activator (tPA) treatment.53) The use of tPA, given at 6h after the thrombosis, causes cerebral hemorrhage at the ipsilateral side of the cerebral cortex and striatum, but stable and long-lasting hyperalgesia was observed bilaterally. The hyperalgesia on both sides was abolished in LPA1 and LPA3 receptor-KO mice, and the chronic pain was cured by repeated treatments with Ki61425, an LPA1/3 receptor antagonist. Although detailed mechanisms underlying bilateral hyperalgesia remain elusive, it may be related to the tPA-specific LPA production at the medial dorsal thalamus, which mediates emotional central pain.54,55) Thus, LPA1 and LPA3 receptor signaling were found to play roles in the development and maintenance of peripheral and central neuropathic pain.
WHO has recently released the new International Classification of Diseases, ICD-11 (May 25, 2019), where fibromyalgia has been classified into a new group, Chronic Primary Pain, while neuropathic pain, chronic cancer-related pain, and other types of pain are classified as Chronic Secondary Pain, since they are at least initially conceived as a symptom. Fibromyalgia is a generalized or widespread pain, found predominantly in females. In developed countries, fibromyalgia is known to comprise approximately 2% of the population.56,57) The ratio of female fibromyalgia patients to males is reported as 8-9 : 1 in western countries, while in Japan it is 4-5 : 1. Unlike neuropathic pain, the cause has not been determined, but emotional stress may be one possible cause. In terms of pharmacotherapy, fibromyalgia is resistant to treatment with NSAIDs and morphine, but has sensitivity to the approved medicines pregabalin and duloxetine, which have side effects such as dizziness, drowsiness or peripheral edema. As pregabalin or duloxetine are known to have symptomatic mechanisms through an inhibition of pain transmission or an enhancement of endogenous pain suppression,58,59) they are also presumed to have different sites of action, which unfortunately lead to side effects during long-term treatment.
4.1. Animal Model for FibromyalgiaTo discover radical treatments for fibromyalgia, or centralized pain, which would be more beneficial for individuals with chronic pain disease, the development of experimental animal models would be necessary. Two research groups have been pioneers in developing different types of widespread chronic pain models in rodents. Levine’s group developed vagotomized animals,60) and Sluka et al. developed an acid saline-induced muscle (ASM) pain model.61) Both models showed sensitivities to morphine as well as to gabapentinoids and antidepressants.62–65) We have also developed a new type of widespread pain model by use of intermittent cold stress (ICS) in mice. The ICS-model shares many clinical features of fibromyalgia patients, such as chronic, widespread, female-dominant pain in terms of pathophysiology.66) Moreover, the hyperalgesia in the ICS model also shows resistance to morphine treatment, consistent with clinical fibromyalgia observations.57) Major mechanisms of morphine resistance have been found to be related to a lack of opioid action in the brain, but not in the spinal cord.
4.2. New Animal Model for Fibromyalgia Using EmpathyIn order to confirm whether intermittent (or unexpected) stress is necessary for the development of fibromyalgia-like widespread pain, we attempted to use different types of stress: intermittent psychological stress (IPS) or empathy.67) In this method using a transparent community box, 5 mice were individually put into transparent compartments which gave an electrical foot shock, while 4 other mice were also individually put into compartments without foot shock, as shown in Fig. 7. Accordingly, the 4 mice without foot shock were automatically given indirect psychological stress or empathy by vision, hearing and an unpleasant smell derived from foot-shocked mice. The mice with foot shock (approx. 1 h per day for 5 d) showed a rapid decrease in pain threshold, but the hyperalgesia rapidly disappeared by 6 d after the foot shock stress sessions. On the other hand, mice without foot shock, but with psychological stress, exhibited long-lasting hyperalgesia, for more than 19 d. The empathy-induced hyperalgesia was more efficient when the stress was randomly programmed. As the pain behaviors were observed not only with various types of pain stimuli (thermal, mechanical and electrical) to the paw, but also with mechanical stimuli to the femoral muscle, the nature of pain seems to be widespread. Female predominant pain was observed, but the difference was very limited. We used mice which had been given gonadectomies 3 weeks before the stress. The IPS-induced hyperalgesia in male mice lasted only 8 d, and completely disappeared at day 15. In female mice given the gonadectomy, on the other hand, IPS-induced hyperalgesia lasted throughout the experiments.
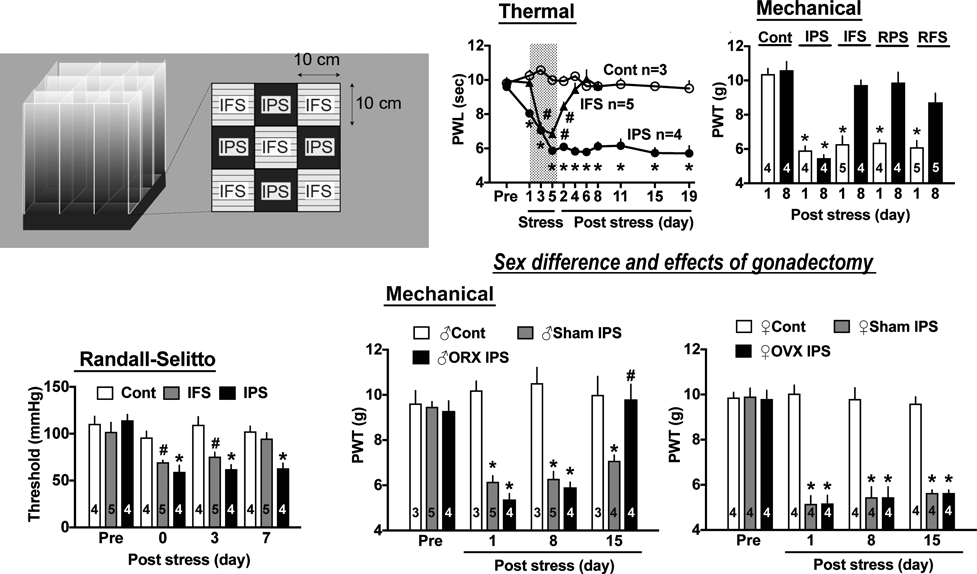
A: Community box for psychological stress. B, C: Intermittent psychological stress-specific long-lasting hyperalgesia. D: Widespread pain evidenced by the hyperalgesia in femoral muscle pain. E, F: Female predominant pain. The sex-difference was more evident in experiments using mice with gonadectomies. Details are described in the text. All data are from an article of Ueda and Neyama (Neurobiol. Pain, 1, 16–25, 2017) with written permission.
As with peripheral and central neuropathic pain models, the IPS-induced hyperalgesia was completely lost in LPA1 receptor-KO mice. Furthermore, the hyperalgesia in the ICS-model using cold stress, and muscle pain in the ASM-model using acid saline, was also abolished in LPA1 receptor-KO mice. The established pain in the IPS-treated mice was abolished by repeated central treatments with the LPA1/3 receptor antagonist, AM966.
In comparing clinical studies, we have determined that the numerous existing reports on treating basic mechanisms of fibromyalgia is not sufficient in the search for meaningful treatment for this type of pain. This may be closely related to the fact that the cause of fibromyalgia is not clear or simple. As often reported or discussed, many different life events, including peripheral injuries and emotional stress, may be among the causes leading to fibromyalgia. From existing clinical and basic reports, however, the primary mechanisms underlying fibromyalgia are presumably in the brain; more specifically, they are closely related to pain memory in the brain. Thus, we propose that fibromyalgia and related pain diseases could be called “centralized pain.” Peripheral abnormalities, including small fiber changes, may be secondary mechanisms which contribute to the reinforcement of centralized pain (Fig. 8). In peripheral neuropathic pain, on the other hand, pain memory should primarily exist in the spinal cord, although brain memory mechanisms would also be working at the same time. As all these mechanisms were blocked by LPA1 receptor- and LPA3 receptor-KO mice or their antagonists, LPA receptor signaling is presumed to contribute to pain memory and/or related neural plasticity. The most important result from our studies on chronic pain is that pain memory is maintained by a feed-forward system. One of the examples of this feed-forward system was observed in the LPA-induced amplification of LPA production. In the case of peripheral nerve injury, local immune cells stimulate sensory fibers, which may also reinforce pain memory in the spinal cord. In central neuropathic pain or centralized pain disease, including fibromyalgia, a big reinforcement loop through neuroimmunological, neuroendocrinological or sympathetic activities may contribute to the feed-forward pain memory system. Finally, I strongly propose that recognition of a “systems pathology of chronic pain” is very important, not only for a better understanding of chronic pain, but also as it may lead to translational research to design new radical treatments.
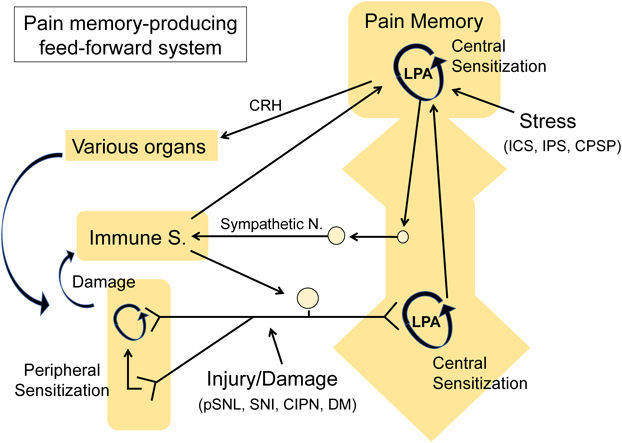
Details are described in the text. Abbreviations: CRH: corticotropin releasing hormone; ICS: intermittent cold stress; IPS: intermittent psychological stress; CPSP: central post stroke pain; pSNL: partial sciatic nerve ligation; SNI: spared nerve injury; CIPN: chemotherapy induced peripheral neuropathy; DM: Diabetes Mellitus. (Color figure can be accessed in the online version.)
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant number 26253077 to Hiroshi Ueda) and by the Platform for Drug Discovery, Informatics, and Structural Life Science [16am0101012j0005] (H.U.) from the Japan Agency for Medical Research and Development (AMED).
The author declares no conflict of interest.
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award.