Abstract
Endoplasmic reticulum (ER) stress-mediated apoptosis pathway is considered to play a vital role in mediating stroke and other cerebrovascular diseases. Previous studies have showed that vascular endothelial growth factor (VEGF) antagonism reduced cerebral ischemic–reperfusion (CI/R) damage, but whether attenuation of ER stress-induced apoptosis is contributing to its mechanisms remains elusive. Our study aimed to investigate the protective effect of VEGF antagonism on CI/R-induced injury. First, oxygen–glucose deprivation and re-oxygenation (OGD/R) BEND3 cell model was constructed to estimate small interfering RNA (siRNA)-VEGF on damage of endothelial cells. Next, in animal model, CI/R mice were induced by middle cerebral artery occlusion (MCAO) for 2 h followed by 24 h reperfusion to investigate cerebral tissue damage. For treatment group, mice received 100 µg/kg anti-VEGF antibodies at 30 min before MCAO, followed by 24 h reperfusion. Our findings demonstrated that pre-administration of siRNA-VEGF before OGD/R changed the biological characteristics of BEND3 cells, reversed the levels of X-box binding protein-1 (XBP-1) and glucose-regulated protein 78 (GRP78), showing siRNA-VEGF attenuated, at least in part, the oxidative damage in OGD/R cell by down-regulating ER stress. In mice experiment, pre-administration of anti-VEGF antibody reduced the brain infarct volume and edema extent and improved neurological scores outcome of CI/R injury mice. Pathological and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining results also confirmed this protective effect. The expressions of VEGF, CATT/EBP homologous protein (CHOP), inositol requiring enzyme 1α (IRE-1α), and cleaved-caspase12 and c-jun N-terminal kinase (JNK) phosphorylation were also prominently decreased. These results suggested that inhibition of endogenous VEGF attenuates CI/R-induced injury via inhibiting ER stress-mediated apoptosis.
INTRODUCTION
As one of the most damaging neurological disease, cerebral stroke affects approximately 3 million people in china and this data is showing a continuous upward trend.1) Cerebral stroke usually causes severe cranial nervous system dysfunction and even loss of mobility and independence, further leading to the family’s economic and psychological burden.2) In nature, ischemic stroke is the most common, which is the result of thrombosis or embolism leading to the immediate loss of oxygen and glucose in specific brain regions.3) In theory, significant neurological and functional deficits can be prevented if measures are taken within critical time windows.4) However, although the recovery of blood circulation in ischemic brain tissue is a necessary condition for patients to return to normal function, brain injury after cerebral ischemia–reperfusion (CI/R) is still inevitable.5) CI/R injury usually causes malignant cerebral edema, affecting neurons and ultimately leading to cell death, however the underlying mechanism of CI/R injury-induced cell death remains to be elucidated.
In cell microenvironment, endoplasmic reticulum (ER) plays an important role in the process of secreting proteins and the membrane proteins folding. The pathological state of ER stress will occur when ER function is impaired.6) ER stress leads to the accumulation of unfolded proteins in the ER, and long-term or excessive accumulation can cause unfolded protein response, which in turn induce apoptosis.7) As one of dangerous biochemical events triggered by cerebral ischemia, ER stress-mediated apoptosis ultimately lead to the death of brain cells.8) ER stress induced by CI/R injury is usually associated with the increased expressions of ER stress markers such as CAT T/EBP homologous protein (CHOP), X-box binding protein-1 (XBP-1), and glucose-regulated protein 78 (GRP78).9) Meanwhile, elevated levels of calcium (Ca2+) and reactive oxygen species (ROS) also cause differential expression of these markers.10) Many studies have shown that ER stress is a key component of the pro-apoptotic signaling pathway.11) When the duration of ER stress is too long or too strong, its homeostasis cannot be re-established, and the apoptotic effector molecules transduced by inositol requiring enzyme 1 (IRE1) pathway can induce apoptosis through the activation of c-Jun N-terminal kinase (JNK) and caspase-12, and CHOP transcriptional up-regulation.12,13) Moreover, recent evidences have indicated that ER stress plays a key role in brain injury following CI/R injury, and that this type of damage can be alleviated via inhibiting endoplasmic reticulum-induced apoptosis by various means.14,15) Hence, how to effectively inhibit the ER stress induced by CI/R injury will provide novel ideas for the treatment of stroke and other diseases.
Disruption of the blood–brain barrier (BBB), one of the predictable consequences of cerebral ischemia, has been proved to be related not only to degeneration of brain function, angioedema, but also to cellular edema and neuronal death.16) Vascular endothelial growth factor (VEGF) is one of the potent mediators for maintaining vascular permeability.17) In the case of general cerebral ischemia, the expression of VEGF and its receptors in the blood vessels, neurons, and other cells is increased significantly.18) For example, intravenous administration of VEGF to rats 1 h after the onset of ischemia significantly exacerbated BBB leakage in the ischemic area.19) Conversely, inhibition of VEGF has been successfully reduced in vascular regression and permeability, and eventually successfully resolved in tumors.20,21) In clinical study, elevated VEGF levels can be detected as early as 3 h after stroke. Inhibition of the VEGF pathway by binding to the VEGF receptor (VEGFR) can target VEGF, which has a preclinical benefit in cerebrovascular disease.22,23) In animal model, VEGF antagonism reduces edema formation and tissue damage after CI/R injury in mouse.24) All of these observations showed VEGF has become an interesting potential therapeutic target preventing brain tissues damage and neuronal cell death after cerebral ischemia.
In this study, we assumed that inhibition of endogenous VEGF in CI/R rat model or oxygen–glucose deprivation and re-oxygenation (OGD/R) cell model would attenuate the cell and tissue injury induced by ER stress. Our results suggest that VEGF antagonism attenuates ER stress-dependent apoptotic signaling pathways, thereby protecting CI/R mice and OGD/R cells, suggesting that it can be used as a potential treatment for ischemic stroke.
MATERIALS AND METHODS
Cell Culture and AnimalsMouse brain microvascular endothelial cell BEND3 (#CRL-2299) was obtained from American Type Culture Collection (ATC C). BEND3 was cultured in Dulbecco’s modified Eagle’s medium (DMEM, #30-2002, ATC C) supplemented with 10% fetal bovine serum (FBS, #30-2020, ATC C) and antibiotics (penicillin, 100 U/mL and streptomycin, 100 µg/mL). Culture system is a humidified atmosphere of 5% CO2/95% air at 37°C. Sixty adult male BALB/C mice (body weight, 18 to 23 g) were purchased from Beijing Hua Fukang Bioscience Co., Inc. (Beijing, China). All mice were maintained on a 12 h light/dark cycle in a temperature room at 22 to 25°C and allowed free access to food and water before surgery.
Construction of Oxygen–Glucose Deprivation and Re-oxygenation (OGD/R) Model and Cell TransfectionTo mimic ischemic condition in vitro, well-grown BEND3 (1 × 104 cells/well) was exposed to oxygen and glucose deprivation (OGD) as we described previously.25) In breif, the cultures were then incubated with a glucose-free Earle’s balanced salt solution (containing a deoxygenated reagent, 0.5 mmol/L sodium dithionite) and immediately transferred to a humidified anaerobic chamber (Reming Bioinstrument, Redfield, NY, U.S.A.) perfused with 95% N2 and 5% CO2 at 37°C for 2 h. For treatment groups, the cells were transfected with small interfering RNA (siRNA)-VEGF (Genscript, Nanjing, China) or siRNA-negative control (NC) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer’s instructions for 6 h before OGD. Afterwards, the medium was replaced by growth medium containing 5.0 mg/mL glucose and cells were cultured in a normoxic incubator with 95% air and 5% CO2 for re-oxygenation of 24 h. Cells cultured in normal medium and normoxic conditions served as a control.
Cell Viability AssayThe cell viability was determined by methyl thiazolyl tetrazolium (MTT) assay according to manufacturer’s instructions. After re-oxygenation of 24 h, 5 mg/mL MTT was added to each well, followed by incubation for 4 h at 37°C. The medium was then removed, and dimethylsulfoxide was added to each well. The absorbance at the 490 nm wavelength was measured using iMark microplate reader (Bio-Rad, CA, U.S.A.). Cell viability was calculated as follows: cell viability (%) = [(treated group A value−untreated control A value)]/[(control group A value−untreated control A value)] × 100%.
Apoptosis AssayAfter re-oxygenation of 24 h, each group of cells was fixed in ice-cold 75% ethanol for 2 h, washed and centrifuged at 1500 rpm for 10 min, and then cells were harvested at a density of 5 × 105 cells/mL for the apoptosis assay by using the Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) double-labelled flow cytometry kit (#CA1020, Solarbio, Beijing, China). At last, data were acquired by a FC500 MCL flow cytometer (Beckman, U.S.A.) to evaluate the apoptotic rate (B2 + B4 quadrant) of each sample.
Cell Cycle AssayAfter re-oxygenation of 24 h, each group of cells was fixed in ice-cold 75% ethanol for 2 h, washed and centrifuged at 1500 rpm for 10 min, and then cells were harvested at a density of 5 × 105 cells/mL and labelled with PI from PI staining cell cycle detection kit (#KGA512, Keygen, Jiangsu, China). At last, flow cytometry assays for cell cycle of each group were carried out with a FC500 MCL flow cytometer to analyze the changes of cell cycle.
Detection of Reactive Oxygen Species (ROS)Intracellular ROS level was measured using 2′,7′-dichlorodihydroluorescein diacetate (DCFH-DA, #E004, Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). After re-oxygenation of 24 h, each group of cells was washed and centrifuged at 1500 rpm for 10 min with phosphate buffered solution (PBS), and then cells were harvested at a density of 1.0 × 106 cells/mL labeled with 10 µM DCFH-DA for 30 min at 37°C. With that, excess DCFH-DA was removed by washing the cells in serum-free DMEM medium. At last, a FC500 MCL flow cytometer measured the fluorescence intensities.
Enzyme-Linked Immune Sorbent Assay (ELISA) for VEGFAfter re-oxygenation of 24 h, the culture supernatant of each group of cells was collected. VEGF contents in cell culture supernatants were assessed using a VEGF mouse ELISA kit (#EMVEGFACL) from Invitrogen following by manufacturer’s instructions. And then, absorbance was detected with a microplate reader (Bio-Rad) at 450 nm. Finally, the value of various indicators was calculated by a standard curve.
Quantitative Real-Time PCRTotal RNA isolation and quantitative real-time PCR (qPCR) analysis were performed to evaluate the expression changes. Total RNAs of cells were isolated using Trizol (Invitrogen) and they were reverse transcribed into cDNA using the PrimeScript RT kit (TaKaRa Biotechnology, Dalian, China) according to the manufacturer’s protocol. Then qRT-PCR was performed in a 20 µL reaction mixture containing SYBR Green PCR Master Mix (TaKaRa Biotechnology), cDNA and 0.2 mmol/L of each primer at 95°C for 10 min, 40 cycles at 95°C for 10 s and 60°C for 45 s. The data were collected using the QuantStudio™ 6 Flex Real-Time PCR System (Applied Biosystems). The relative amount of each gene was normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Relative quantity values were analyzed using the 2-ΔΔCt method. Primer sequences were shown as following: VEGF-forward 5′-GTC CGA GCC GGA GAG GGA GC-3′, VEGF-reverse 5′-TCT GTC GTG GGT GCA GCC TG-3′; XBP-1-forward 5′-ACA GAG TAG CAG CGC AGA CT-3′, XBP-1-reverse 5′-CTC TAA AAC TAG AGG CTT GG-3′; GRP78-forward 5′-CAT GGT TCT CAC TAA AAT GA-3′, GRP78-reverse 5′-GCT GGT ACA GTA ACA ACT G-3′; GAPDH-forward 5′-TGT GTC CGT CGT GGA TCT GA-3′, GAPDH-reverse 5′-TTG CTG TTG AAG TCG CAG GAG-3′.
Animal Experimental Grouping, CR/I Model Building and Drug AdministrationMice were randomly divided into Sham group (n = 15), CR/I model group (n = 15), CR/I plus rabbit immunoglobulin G (IgG) isotype control antibody group (IC-IgG Anb, #30000-0-AP, Proteintech Group, Inc., Chicago, IL, U.S.A., n = 15) and CR/I plus anti-VEGF Anb group (anti-VEGF Anb, #19003-1-AP, Proteintech Group, Inc., n = 15). For model group, mice underwent middle cerebral artery occlusion (MCAO) surgical operation to induce focal cerebral ischemia. Ischemia and reperfusion conditions were confirmed by the regional cerebral blood flow detected by a laser Doppler cerebral flow meter (PeriFlux 5000, Perimed, Sweden) before cerebral ischemia, during cerebral ischemia, and after reperfusion. When the blood flow volume drops to 50 µL/kg/min, blockage of cerebral blood flow was confirmed. After MCAO for 2 h, 24 h reperfusion was achieved by careful suture removal. However, the mice in the Sham group underwent the same surgical procedure without occlusion. For drug treatment groups, mice received IC-IgG Anb (100 µg/kg) or anti-VEGF Anb (100 µg/kg) by intraperitoneal administration 30 min before MCAO, followed by 24 h reperfusion. Prior to subsequent experiments, mice in each subgroup were sacrificed and brain tissues were collected for the following analysis. The Animal Committee of the First People’s Hospital of Yunnan Province approved all animal experimental protocols and animal handling procedures.
Neurological Score AssessmentAfter 24 h reperfusion, 8 mice were randomly selected from each group for testing. The neurological scores of each group of were measured using Garcia’s neurological score method by a single experimenter who was blinded to the experimental treatment groups. The behavior of each mouse was assessed using six independent criteria: (1) spontaneous activity, (2) symmetry of movements, (3) outstretching of the forelimbs, (4) wall climbing, (5) reaction to body touch, and (6) response to touching of the vibrissae. Of the six parameters, the first three have a minimum score of 1, and the remaining three have 0 as their minimum score. In addition, all of the six parameters have a maximum score of 3; Thus, the total score for each mouse ranged from 3 to 18 points.26)
2,3,5-Triphenyltetrazolium Chloride StainingIn order to assess the volumes of the infarcts, we selected 4 mice from each group randomly for decapitation, 24 h after the post-ischemic reperfusion followed by MCAO. And then brains were collected for 2,3,5-triphenyltetrazolium chloride (TTC, #T8877, Sigma) staining. The color of normal brain tissue is evenly red, while the infarcted area is whitish. The ratio of infarct volume out of the entire brain represented the degree of cerebral infarction and was analyzed using the Adobe Photoshop CS imaging system.
Extent of Brain Edema MeasurementIn order to assess brain water content, we selected 3 mice from each group randomly for decapitation, 24 h after the post-ischemic reperfusion followed by the MCAO. In this process, the right hemisphere was quickly separated from the entire brain. Each sample was dried in an oven at 100°C for 24 h after being weighed on an electronic balance. Next, weigh each sample again and calculate the water content using the following formula: [(wet weight minus dry weight)/wet weight] × 100%. Meanwhile, the remaining left brain tissue was immobilized or frozen for subsequent molecular detection.
Haematoxylin and Eosin (H&E) and Nissl StainingPre-fixed brain tissue was selected, embedded with paraffin, cut into 4–5 µm thick sections, and stained by H&E staining for evaluating the structural changes or Nissl staining for assessing the injury of neurons. Stained slides were captured with the microscope (Olympus, Tokyo, Japan). The extent of tissue disorganizations and neurons injury were noted for each sample.
Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate Nick-End Labeling (TUNEL) StainingTUNEL staining was performed using a situ TUNEL apoptosis detection kit (#G002-1, Nanjing Jiancheng Bioengineering Institute, Jiangsu, China) according to the manufacturer’s instructions. Briefly, paraffin-embedded sections were deparaffinized and rehydrated, and then dealed with proteinase K (10 µg/mL) at 37°C for 30 min. The sections were washed in PBS for 3 times and then incubated in the 50 µL TUNEL reaction buffer including the enzyme at 37°C for 30 min away from light. Finally, the samples were counterstained with 4′-6-diamidino-2-phenylindole (DAPI) (Sigma) to visualize cell nuclei and photographed using a fluorescence microscope (Leica, Biosystems, Germany). Count positive and total cells in three fields at 400 magnification by blinded technicians.
Protein Extraction and Western Blot AnalysisThe total proteins of each group cell and brain tissue were extracted using corresponding extraction kit and quantified by the total protein assay kit (BCA method). With that, whole proteins were electrophoresed under the conditions in 12% polyacrylamide gels. The separated proteins were transferred to polyvinylidene difluoride membranes. Nonspecific binding was blocked with 5% skimmed milk at 37°C for 2 h. And then, incubate the membranes using the listed primary antibody: rabbit anti-Cleaved-caspase3 (#ab49822, 1 : 1000, Abcam), rabbit anti-Cyclin D1 (#ab134175, 1 : 1000, Abcam), rabbit anti-VEGF (#19003-1-AP, 1 : 1000, Proteintech Group, Inc.), rabbit anti-CHOP (#ab10444, 1 : 1000, Abcam), rabbit anti-IRE1α (#3294, 1 : 1000, CST), rabbit anti-JNK (#ab179461, 1 : 1000, Abcam), rabbit anti-p-JNK (#ab124956, 1 : 1000, Abcam), rabbit anti-Total caspase12 (#ab62484, 1 : 1000, Abcam), rabbit anti-Cleaved-caspase12 (#ab8118, 1 : 1000, Abcam), rabbit anti-GAPDH (#ab181602, 1 : 1000, Abcam). Next, the membranes were incubated with goat anti rabbit secondary antibody HRP-conjugated IgG (#ab6721, 1 : 5000, Abcam) for 1 h at room temperature, and finally developed with diaminobenzidine tetrahydrochloride (DAB). At last, images were obtained from multifunctional Gel Imaging System (Image Quant LAS 500, General Electric, Fairfield, CT, U.S.A.).
Statistical AnalysesAll values are presented as the means ± standard deviation (S.D.) Differences in multiple groups (more than 2) were compared using SPSS 19.0 software (IBM Corp., Armonk, NY, U.S.A.) using one-way ANOVA followed by Tukey’s post hoc test. The neurological score data were analyzed by a Mann–Whitney U-test. p < 0.05 was considered to indicate a statistically significant difference.
RESULTS
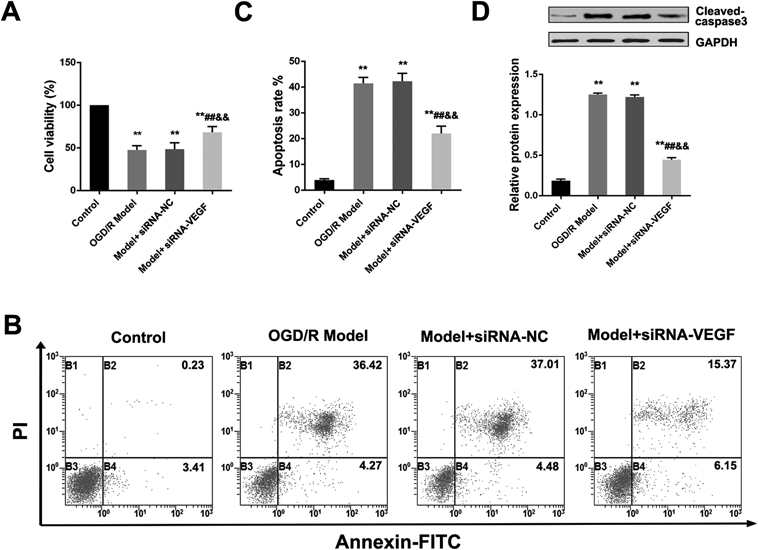
VEGF Inhibition Reversed Proliferation Activity and Inhibited Apoptosis of OGD/R Model CellAt 24 h after re-oxygenation, OGD/R injury significantly reduced BEND3 cell viability when compared with control group (p < 0.01). Nevertheless, compared with OGD/R model group and OGD/R model plus siRNA-NC group, VEGF inhibition dramatically increased BEND3 cell viability (all p < 0.01) (Fig. 1A). As the apoptosis test results shown in Figs. 1B and C, VEGF inhibition significantly suppressed the apoptosis of OGD/R induced BEND3 cell when compared with model group (p < 0.01) or OGD/R model plus siRNA-NC group (p < 0.01). In addition, apoptosis-related protein Cleaved-caspase3 level was significantly reduced with siRNA-VEGF pretreatment (Fig. 1D). These results demonstrated that VEGF inhibition with siRNA-VEGF not only reverses proliferation activity, but also inhibits apoptosis of OGD/R model cell.
VEGF Inhibition Suppressed G0/G1 Phase BEND3 Cell Cycle Arrest Induced by OGD/RAt 24 h after oxygenation, the cell cycle of the OGD/R model group was arrested obviously in the G0/G1 phase contrasted with the control group. However, compared with OGD/R model group and OGD/R model plus siRNA-NC group, VEGF inhibition clearly reversed the blockage of OGD/R on the cell cycle (all p < 0.01) (Figs. 2A, B). At the same time, the expression of cell cycle regulated-protein Cyclin D1 was re-increased by siRNA-VEGF pretreatment from a low expression state (Fig. 2C). Thus it can be seen that VEGF inhibition with siRNA-VEGF can suppress G0/G1 phase BEND3 cell cycle arrest induced by OGD/R, which may be achieved through the regulation of Cyclin D1 protein expression.
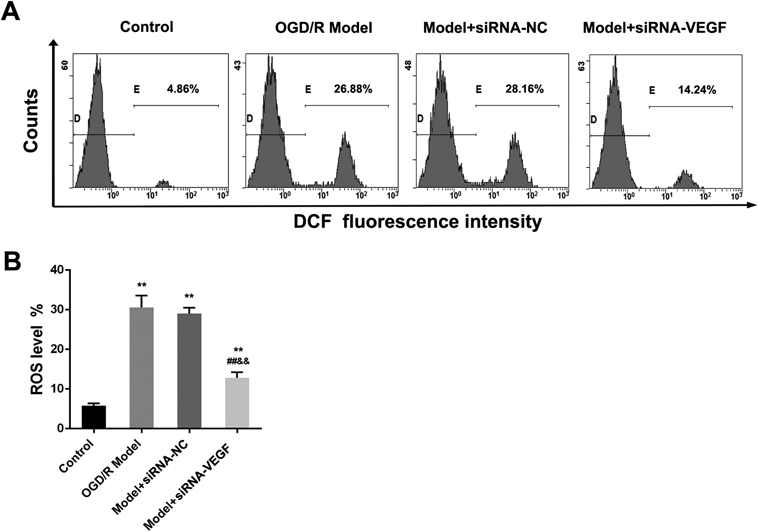
VEGF Inhibition Down-Regulated the ROS Level of BEND3 Cell Induced by OGD/RAt 24 h after re-oxygenation, oxidative stress phenomenon in each group cell was quantified by detecting ROS level. As shown in Figs. 3A and B, OGD/R significantly increased the level of ROS level in BEND3 cell compared with control group (p < 0.01). However, compared with OGD/R model group and OGD/R model plus siRNA-NC group, VEGF inhibition clearly decreased the ROS level of OGD/R induced BEND3 cell (all p < 0.01). VEGF inhibition with siRNA-VEGF may slow down the occurrence of oxidative stress by reducing ROS levels in BEND3 cell induced by OGD/R.
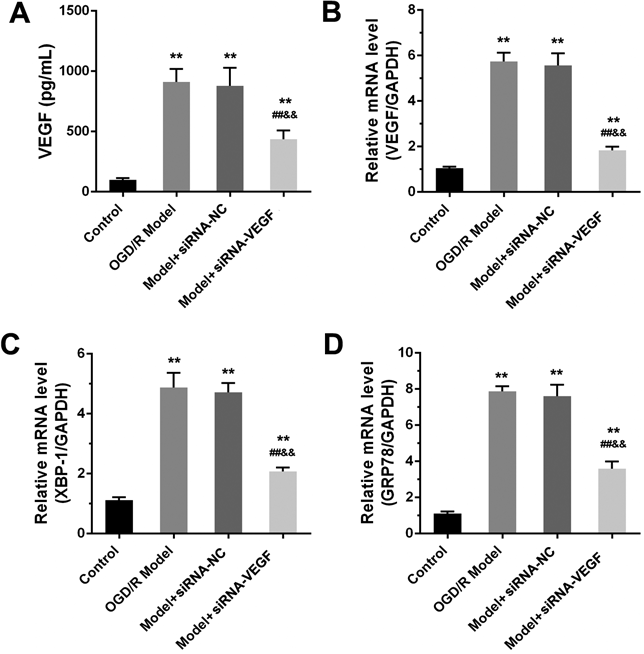
VEGF Inhibition Decreased the Level of VEGF and ER Stress Markers in OGD/R Model CellAt 24 h after re-oxygenation, cell culture supernatants were collected and used for VEGF secretion level detection by ELISA. Meanwhile, the centrifuged and washed cells were extracted RNA for qRT-PCR. On the one hand, as shown in Figs. 4A and B, OGD/R significantly increased the secretion level and mRNA level of VEGF in BEND3 cell compared with control group (all p < 0.01). Conversely, VEGF inhibition with siRNA-VEGF clearly decreased the secretion level and mRNA level of VEGF when compared with OGD/R model group and OGD/R model plus siRNA-NC group (all p < 0.01). On the other hand, as shown in Figs. 4C and D, siRNA-VEGF pretreatment also significantly decreased the mRNA level of ER stress markers (XBP-1 and GRP78) when compared with OGD/R model group and OGD/R model plus siRNA-NC group (all p < 0.01). All of those results demonstrated that the repair effect of VEGF inhibition with siRNA-VEGF on BEND3 cell injury induced by OGD/R may be closely related to inhibition of ER stress occurrence.
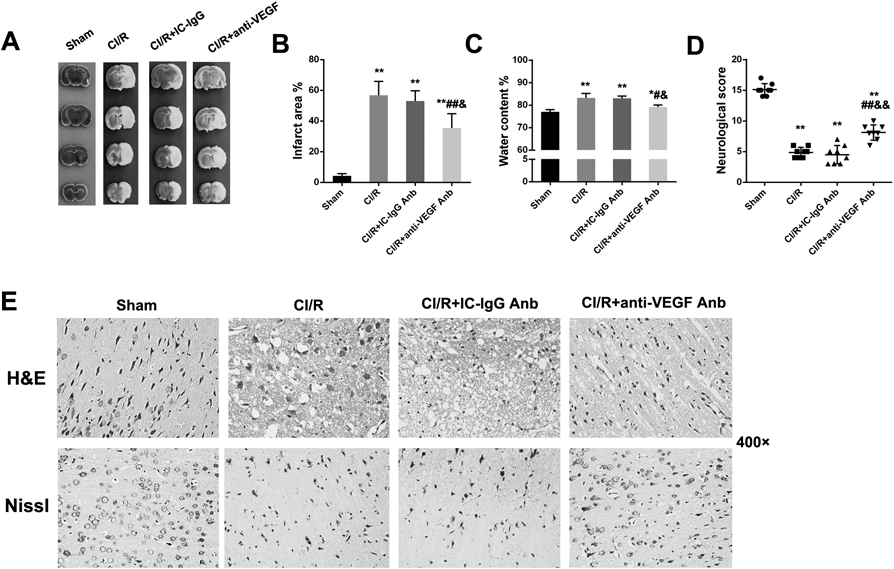
VEGF Antagonism Reduced the Brain Infarct Volume and Edema Extent and Improved Neurological Scores Outcome of CI/R Injury MiceWe measured the infarct volume by TTC staining in brain sections to confirm the neuroprotective effect of VEGF antagonism on cerebral ischemic injury. The results showed that VEGF antagonism resulted in a reduction in infarct size, compared with the CI/R model group and CI/R plus IC-IgG Anb group (p < 0.05, p < 0.01) (Figs. 5A, B). As shown in Fig. 5C, anti-VEGF Anb treatment also significantly decreased the brain edema extent when compared with CR/I model group and CI/R plus IC-IgG Anb group (all p < 0.05). In addition, it was shown that the neurological function of the mice receiving VEGF antagonism was significantly improved (p < 0.01) by measuring the behavioral function scores of mice in different groups 24 h after reperfusion. (Fig. 5D). Thus, these results demonstrated that anti-VEGF Anb shows a significant protection on brain injury of CI/R mice.
VEGF Antagonism Improved Brain Histopathological Changes and Neuronal Injury of CI/R MiceAs shown in Fig. 5E, H&E staining results showed more neurons in the cerebral cortex of sham group mice, with clear morphological structure, regular cell arrangement and deep cell nucleus staining and obvious nucleoli. In the cerebral cortex of the CI/R model group and CI/R plus IC-IgG Anb group, the ethmoid softening nerve cells were observed, the glial cells were edematous and the cytoplasm was loose. However, nerve cells in the VEGF antagonism group and the sham group had the similar morphology, showing a relatively normal structure with marked reduction in interstitial edema. Meanwhile, Nissl staining results showed no morphological changes of neurons in cortex in the sham group. In the CI/R model group and CI/R plus IC-IgG Anb group, most neurons showed that Nissl’s body disappeared, the chromosomes condense, and the nucleus shrank or the cellular structure disappear. Interestingly, degenerated neurons in mice pretreated with anti-VEGF Anb were reduced, and the amount of intact neurons was increased obviously. All results demonstrated that VEGF antagonism improves brain histopathological changes and neuronal injury of CI/R mice.
VEGF Antagonism Decreased the Apoptotic Cells in Brain of CI/R Injury MiceThe apoptotic cell death in brain tissues was determined by TUNEL staining, and the results were shown in Fig. 6A. Damaged brain sections showed TUNEL positive cells, while no significant TUNEL positive cells were detected in the Sham group. In CI/R model group, the TUNEL-positive cells were observed obviously, and showed a significant difference compared to sham group (p < 0.01). On the contrary, pre-treatment of VEGF antagonism significantly reduced the number of TUNEL-positive cells compared to the CI/R model group and CI/R plus IC-IgG Anb group (all p < 0.01) (Fig. 6B).
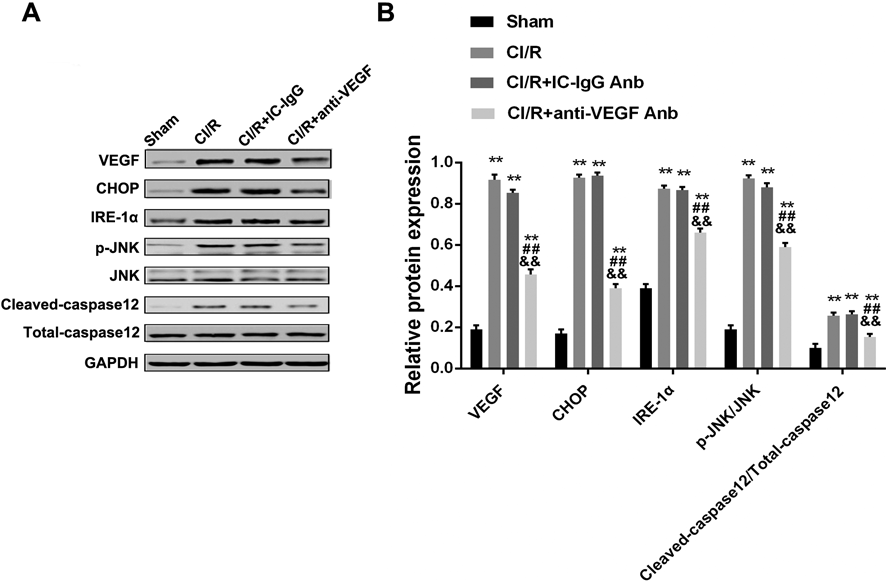
VEGF Antagonism Suppressed ER Stress Mediated-Apoptosis PathwayAs demonstrated in Fig. 7, CI/R induced up-regulation of VEGF, CHOP and IRE-1α protein levels, phosphorylation of JNK and cleaved caspase-12 levels in injury mice cerebral tissues, suggesting that ER stress and it mediated-apoptosis pathway were triggered in CI/R-induced mice. However, pre-treatment with anti-VEGF Anb effectively modulated ER stress activation down to near normal. In brief, the results of differential protein levels showed that VEGF antagonism suppress ER stress mediated-apoptosis pathway.
DISCUSSION
The major findings of the present study suggested that siRNA-VEGF pretreatment provided a protective effect for OGD/R induced BEND3 cell by inhibiting ROS level, regulating cell cycle and arresting apoptosis, and further enhancing cell viability. Furthermore, this study also showed in vivo that pretreatment of VEGF antagonism protects against CI/R induced cerebral injury by decreasing ER stress-mediated apoptosis.
Stroke is generally considered to induce neuronal damage and further exasperate brain injury leading to the development of limb dysfunction, which is tight junction with the destruction of the BBB and characterized by the blood crossing the blood–brain barrier and overflowing directly into the brain parenchyma.27) However, dysfunction of endothelial growth cell may be an important pathophysiological factor in stroke or others human cerebrovascular diseases.28) As a potent mediator of vascular permeability, binding of VEGF to VEGF receptors results in cell survival, vascular permeability mediated, endothelial cell differentiation and other impacts. An interesting study showed that the destruction of BBB is aggravated by exogenous VEGF in the early stage of focal ischemia.29) In ischemic rat model, BBB disruption and ischemic lesions were also significantly increased by early post-ischemic administration of recombinant human VEGF 165.23) In contrast, a significant attenuation of BBB disruption was detected with anti-VEGF antibody application in the early period of focal ischemia, however no specific mechanism was shown.19) Thus, we investigated the feasibility and mechanism of VEGF inhibition treatment of CI/R injury in vitro and in vivo in depth.
OGD/R cell model can well simulate ischemia reperfusion injury in vitro. Our data clearly showed that pre-administration of siRNA-VEGF before OGD/R could change the biological behaviors of BEND3 cells. In addition, molecular detection results showed that the secretion level and mRNA level of endogenous VEGF in OGD/R cell is significantly reduced. And the levels of ER stress markers (XBP-1 and GRP78) were reversed by siRNA-VEGF from a relatively high expression level. Former researches have shown that ROS is closely related to ER stress, as the increase in protein folding results in the accumulation of ROS.30,31) These findings suggested that this protective mechanism may inhibit the production of intracellular ROS via suppressing the endogenous VEGF expression in OGD/R cell. Besides, it could be assumed from decreased levels of ER stress markers that siRNA-VEGF pretreatment attenuated, at least in part, the oxidative damage in OGD/R cell by down-regulating ER stress response.
In our in vivo experiment, the generally accepted CI (MCAO)/R model was chosen for this research. As we predicted, pre-administration of anti-VEGF antibody reduced the brain infarct volume and edema extent and improved neurological scores outcome of CI/R injury mice. Pathological staining results also confirmed the protective effect of anti-VEGF antibody on brain injury of CI/R mice. Apoptosis is one of the major cell death patterns in ischemic penumbra, particularly induced by the ER stress pathway.15) In our findings, apoptotic neurons of brain sections were identified by TUNEL staining, showing a significantly reduced number of TUNEL-positive cells in the anti-VEGF antibody treatment group than in the CI/R model group. These results indicated that the positive effects of anti-VEGF antibodies are partially related to their anti-apoptotic effects at any rate. For this series of phenomena, we believe that anti-VEGF antibody neutralizes the high level of endogenous VEGF at the time of CI/R injury. The study of Chi et al. has also shown that inhibition of endogenous VEGF by topical application of anti-VEGF antibody in the ischemic cortex decreased cerebral ischemia injury, suggesting that endogenous VEGF is in part responsible for the BBB disruption during the early stage of focal cerebral ischemia.32) However, to further elucidate the relationship between the effect of anti-VEGF antibodies and neuronal apoptosis on this process, we then examined the effects of anti-VEGF antibody on molecular events related to ER stress mediated-apoptotic pathways.
As one of the classic pathways of apoptosis, ER stress-mediated apoptosis has been shown to be participate in various diseases and can be regard as a potential target for multiple drugs.33) For example, in Parkinson’s disease research, ER stress-mediated apoptosis is involved in manganese-induced neurotoxicity, demonstrated by increased expressions of p-IRE1, ATF6, PERK, GRP78, CHOP, caspase-12 and caspase-3, and decreased Bcl-2 expression in rat striatal neurocytes.34) In streptozotocin-induced diabetic mice, there were increased protein expressions of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits, GRP78 and ER-associated apoptosis proteins.35) As well, gypenoside inhibited ER stress and apoptosis in ischemic reperfusion injured rats, which was proved to be related the blockade of CHOP pathway and activation of Phosphoinositol 3-kinase/Akt pathway.36) In ER stress-induced apoptosis, the pro-apoptotic transcription factor CHOP also plays a key role, and cells lacking CHOP are significantly resisting the lethal ER stress.37) JNK is also called stress-activated protein kinase (SAPK), and is involved in stress reactions and cell death.38) However, in our study, compared with model group, pre-administration of anti-VEGF antibody prominently decreased the expressions of CHOP, IRE-1α, and cleaved-caspase12 and phosphorylation level of JNK. All the data indicated that this protective mechanism of anti-VEGF antibodies on brain damage may be through inhibition of ER stress-mediated apoptotic pathway.
In conclusion, our study demonstrated that the application of siRNA-VEGF or anti-VEGF antibody to inhibit endogenous VEGF expression can attenuate cerebral I/R-induced injury in vitro and in vivo via depressing ER-stress-mediated apoptosis. VEGF antagonism may be considered as an effective treatment strategy for stroke in the future.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Jiang G, Li W, Wang D, Shen C, Ji Y, Zheng W. Epidemiological transition and distribution of stroke incidence in Tianjin, China, 1988-2010. Public Health, 131, 11–19 (2016).
- 2) Shinohara Y. Hemorrhagic stroke syndromes: clinical manifestations of intracerebral and subarachnoid hemorrhage. Handb. Clin. Neurol., 93, 577–594 (2008).
- 3) Adams HP Jr, del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, Grubb RL, Higashida RT, Jauch EC, Kidwell C, Lyden PD, Morgenstern LB, Qureshi AI, Rosenwasser RH, Scott PA, Wijdicks EF, American Heart Association, American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke, 38, 1655–1711 (2007).
- 4) Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, Yoo AJ, Schonewille WJ, Vos JA, Nederkoorn PJ, Wermer MJ, van Walderveen MA, Staals J, Hofmeijer J, van Oostayen JA, Lycklama à Nijeholt GJ, Boiten J, Brouwer PA, Emmer BJ, de Bruijn SF, van Dijk LC, Kappelle LJ, Lo RH, van Dijk EJ, de Vries J, de Kort PL, van Rooij WJ, van den Berg JS, van Hasselt BA, Aerden LA, Dallinga RJ, Visser MC, Bot JC, Vroomen PC, Eshghi O, Schreuder TH, Heijboer RJ, Keizer K, Tielbeek AV, den Hertog HM, Gerrits DG, van den Berg-Vos RM, Karas GB, Steyerberg EW, Flach HZ, Marquering HA, Sprengers ME, Jenniskens SF, Beenen LF, van den Berg R, Koudstaal PJ, van Zwam WH, Roos YB, van der Lugt A, van Oostenbrugge RJ, Majoie CB, Dippel DW, MR CLEAN Investigators. A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med., 372, 11–20 (2015).
- 5) Renú A, Laredo C, Tudela R, Urra X, Lopez-Rueda A, Llull L, Oleaga L, Amaro S, Chamorro A. Brain hemorrhage after endovascular reperfusion therapy of ischemic stroke: a threshold-finding whole-brain perfusion CT study. J. Cereb. Blood Flow Metab., 37, 153–165 (2017).
- 6) Rausch MP, Sertil AR. A stressful microenvironment: opposing effects of the endoplasmic reticulum stress response in the suppression and enhancement of adaptive tumor immunity. Int. Rev. Immunol., 34, 104–122 (2015).
- 7) Bohnert KR, McMillan JD, Kumar A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol., 233, 67–78 (2018).
- 8) Zhao Q, Wang X, Chen A, Cheng X, Zhang G, Sun J, Zhao Y, Huang Y, Zhu Y. Rhein protects against cerebral ischemic/reperfusioninduced oxidative stress and apoptosis in rats. Int. J. Mol. Med., 41, 2802–2812 (2018).
- 9) Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res., 17, 189–202 (2010).
- 10) Sun FC, Shyu HY, Lee MS, Lee MS, Lai YK. Involvement of calcium-mediated reactive oxygen species in inductive GRP78 expression by geldanamycin in 9L rat brain tumor cells. Int. J. Mol. Sci., 14, 19169–19185 (2013).
- 11) Rozpedek W, Nowak A, Pytel D, Diehl JA, Majsterek I. Molecular basis of human diseases and targeted therapy based on small-molecule inhibitors of ER stress-induced signaling pathways. Curr. Mol. Med., 17, 118–132 (2017).
- 12) Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol., 8, e1000410 (2010).
- 13) Roberson EC, Tully JE, Guala AS, Reiss JN, Godburn KE, Pociask DA, Alcorn JF, Riches DW, Dienz O, Janssen-Heininger YM, Anathy V. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-beta release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol., 46, 573–581 (2012).
- 14) Hu YQ, Chen W, Yan MH, Lai JJ, Tang N, Wu L. Ischemic preconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PERK pathway. Eur. Rev. Med. Pharmacol. Sci., 21, 5736–5744 (2017).
- 15) Wu CX, Liu R, Gao M, Zhao G, Wu S, Wu CF, Du GH. Pinocembrin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress induced apoptosis. Neurosci. Lett., 546, 57–62 (2013).
- 16) Chi OZ, Hunter C, Liu X, Weiss HR. Effects of erythropoietin on blood-brain barrier disruption in focal cerebral ischemia. Pharmacology, 82, 38–42 (2008).
- 17) Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner JM. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation, 97, 99–107 (1998).
- 18) Ma Y, Qu Y, Fei Z. Vascular endothelial growth factor in cerebral ischemia. J. Neurosci. Res., 89, 969–978 (2011).
- 19) Chi OZ, Hunter C, Liu X, Weiss HR. Effects of anti-VEGF antibody on blood-brain barrier disruption in focal cerebral ischemia. Exp. Neurol., 204, 283–287 (2007).
- 20) Erber R, Thurnher A, Katsen AD, Groth G, Kerger H, Hammes HP, Menger MD, Ullrich A, Vajkoczy P. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J., 18, 338–340 (2004).
- 21) Chae SS, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, Sorensen AG, Munn LL, Jain RK, Fukumura D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin. Cancer Res., 16, 3618–3627 (2010).
- 22) Kalka C, Takahashi T, Masuda H, Asahara T, Isner JM. Vascular endothelial growth factor (VEGF): therapeutic angiogenesis and vasculogenesis in the treatment of cardiovascular disease. Med Klin (Munich), 94, 193–201 (1999).
- 23) Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood–brain barrier leakage in the ischemic brain. J. Clin. Invest., 106, 829–838 (2000).
- 24) van Bruggen N, Thibodeaux H, Palmer JT, Lee WP, Fu L, Cairns B, Tumas D, Gerlai R, Williams SP, van Lookeren Campagne M, Ferrara N. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J. Clin. Invest., 104, 1613–1620 (1999).
- 25) Wu L, Ye Z, Pan Y, Li X, Fu X, Zhang B, Li Y, Lin W, Li X, Gao Q. Vascular endothelial growth factor aggravates cerebral ischemia and reperfusion-induced blood–brain-barrier disruption through regulating LOC102640519/HOXC13/ZO-1 signaling. Exp. Cell Res., 369, 275–283 (2018).
- 26) Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke, 26, 627–635, discussion, 635 (1995).
- 27) Si D, Li J, Liu J, Wang X, Wei Z, Tian Q, Wang H, Liu G. Progesterone protects blood-brain barrier function and improves neurological outcome following traumatic brain injury in rats. Exp. Ther. Med., 8, 1010–1014 (2014).
- 28) Takizawa S, Nagata E, Nakayama T, Masuda H, Asahara T. Recent progress in endothelial progenitor cell culture systems: potential for stroke therapy. Neurol. Med. Chir. (Tokyo), 56, 302–309 (2016).
- 29) Chi OZ, Hunter C, Liu X, Weiss HR. Effects of VEGF and nitric oxide synthase inhibition on blood-brain barrier disruption in the ischemic and non-ischemic cerebral cortex. Neurol. Res., 27, 864–868 (2005).
- 30) Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci., 8, 213 (2014).
- 31) Kim J, Choi TG, Ding Y, Kim Y, Ha KS, Lee KH, Kang I, Ha J, Kaufman RJ, Lee J, Choe W, Kim SS. Overexpressed cyclophilin B suppresses apoptosis associated with ROS and Ca2+ homeostasis after ER stress. J. Cell Sci., 121, 3636–3648 (2008).
- 32) Chi OZ, Hunter C, Liu X, Weiss HR. Effects of anti-VEGF antibody on blood–brain barrier disruption in focal cerebral ischemia. Exp. Neurol., 204, 283–287 (2007).
- 33) Gwak H, Kim S, Dhanasekaran DN, Song YS. Resveratrol triggers ER stress-mediated apoptosis by disrupting N-linked glycosylation of proteins in ovarian cancer cells. Cancer Lett., 371, 347–353 (2016).
- 34) Wang T, Li X, Yang D, Zhang H, Zhao P, Fu J, Yao B, Zhou Z. ER stress and ER stress-mediated apoptosis are involved in manganese-induced neurotoxicity in the rat striatum in vivo. Neurotoxicology, 48, 109–119 (2015).
- 35) Lakshmanan AP, Thandavarayan RA, Palaniyandi SS, Sari FR, Meilei H, Giridharan VV, Soetikno V, Suzuki K, Kodama M, Watanabe K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci., 44, 627–634 (2011).
- 36) Yu H, Zhang H, Zhao W, Guo L, Li X, Li Y, Zhang X, Sun Y. Gypenoside protects against myocardial ischemia–reperfusion injury by inhibiting cardiomyocytes apoptosis via inhibition of CHOP pathway and activation of PI3K/Akt pathway in vivo and in vitro. Cell. Physiol. Biochem., 39, 123–136 (2016).
- 37) Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab., 9, 474–481 (2009).
- 38) Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim. Biophys. Acta, 1773, 1341–1348 (2007).