Abstract
Previously, we reported that coffee extract and its constituents, caffeic acid (CA) and p-coumaric acid, inhibit infection by the hepatitis C virus (HCV). In the present report, we identified another coffee-related compound, tannic acid (TA), which also inhibits HCV infection. We systematically evaluated which steps of the viral lifecycle were affected by CA and TA. TA substantially inhibits HCV RNA replication and egression, while CA does not. The infectivity of the HCV pretreated with CA or TA was almost lost. Cellular attachment of HCV particles and their interaction with apolipoprotein E, which is essential for HCV infectivity, were significantly reduced by CA. These results indicate that CA inhibits HCV entry via its direct effect on viral particles and TA inhibits HCV RNA replication and particle egression as well as entry into host cells. Taken together, our findings may provide insights into CA and TA as potential anti-HCV strategies.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped, positive-sense, single-stranded RNA virus in the genus Hepacivirus within the Flaviviridae family.1) Approximately 71 million people worldwide are currently infected with HCV.2) Given that chronic HCV infection is a leading cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma,3) it has been recognised as a major global health concern.4) Although novel HCV therapeutics with direct-acting antivirals have led to remarkably improved treatment outcomes,5) problems still persist; high costs in particular restrict most patients’ access to these effective therapeutics.6)
Caffeic acid (CA), a coffee-related organic acid, is a degraded product of chlorogenic acid, abundant in coffee beans. It is involved in the deep aroma, color, and bitterness of coffee. This polyphenol is also contained in a wide variety of other foods, including fruits, vegetables, and grains. For example, red grape juices and wild chokeberries contain 17–30 mg/L7) and 1.41 mg/g8) CA, respectively. It has many beneficial biological effects, including inhibition of cancer cell proliferation and metastasis9,10) and antiviral activities against a variety of viruses.11–21)
Previously, our group has shown that CA inhibits HCV infection and HCV entry into cells,11) but the detailed antiviral mechanism has not yet been defined. In the present study, we investigated the inhibitory effect of another coffee-related compound, tannic acid (TA), on HCV infection and further examined how CA and TA inhibit HCV infection.
MATERIALS AND METHODS
Cells and Cell CultureHuh7.5.1–8, which is derived from Huh7.5.1,22) is a human hepatoma cell line that is highly permissive to HCV infection.23) Huh7.5.1–8-derived OKH-4 is an occludin (OCLN) knockout cell line that is non-permissive to HCV infection because OCLN is essential for HCV infection.24) Both cells were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 0.1 mM non-essential amino acids, 100 units/mL penicillin G, and 100 µg/mL streptomycin sulfate (referred to as “complete medium”).
ReagentsChlorogenic acid and CA were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Methyl 2,5-dihydroxycinnamate and TA were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Purity of CA and TA is ≥98 and 90% (ACS grade), respectively. Kahweol, kahweol palmitate, cafestol, cafestol palmitate, cafestol eicosanate, cafestol stearate, and cafestol acetate were purchased from LKT Laboratories (St Paul, MN, U.S.A.). p-Coumaric acid was purchased from MP Biomedicals (Santa Ana, CA, U.S.A.). D-(−)-Quinic acid was purchased from Alfa Aesar (Haverhill, MA, U.S.A.). Mouse monoclonal antibody (mAb) against HCV core protein (clone 2H9) was described previously.25) Goat polyclonal antibodies (pAb) against apolipoprotein E (ApoE) protein were purchased from Merck Millipore (Burlington, MA, U.S.A.). Mouse mAb against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein (clone 5A12) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Mouse mAb against HCV NS3 protein (clone 8G2) was purchased from Abcam (Cambridge, U.K.).
Viral Stock, Infection, and QuantificationThe HCV-JFH1 strain, an HCV genotype 2a isolate, was prepared from the culture supernatant of Huh7.5.1–8 cells that had been transfected with in vitro-transcribed HCV-JFH1 RNA.23) After serial passages of the HCV-JFH1 in naïve Huh-7.5.1–8 cells, infectious culture supernatants were collected and used as virus stock in this study. Virus titers were determined by fluorescent focus assay as described previously.23) Total RNA was extracted from culture supernatants or cultured cells and purified using the Viral Nucleic Acid Extraction Kit I and Blood/Cultured Cell Total RNA Mini Kit (FAVORGEN Biotech Corp., Pingtung City, Taiwan), respectively. Quantification of HCV-JFH1 RNA was performed by quantitative (q)RT-PCR as described previously.23)
Cell Viability AssayHuh7.5.1–8 cells were seeded at 5 × 104 cells per well in 48-well plates and cultured at 37°C for 1 d. Cells were then incubated in medium containing each coffee-related compound at 37°C for 3 d. After washing twice with phosphate buffered saline (PBS), cells were treated with 500 µL phenol red-free complete medium containing cell counting kit-8 reagent (Dojindo, Tokyo, Japan) at 37°C. After 1 or 2 h, culture supernatants were collected and transferred to 96-well plates. For each well, the absorbance at 450 nm was measured using an iMark microplate reader (BIORAD, Hercules, CA, U.S.A.).
HCV RNA Replication AssayWe used the subgenomic HCV-JFH1 replicon system described previously.23) In brief, cells were seeded at 5 × 104 cells per well in 48-well plates and cultured at 37°C for 1 d. Subgenomic replicon RNA (1 µg each/well) in vitro transcribed from pSGR-JFH1/Luc was transfected into cells using DMRIE-C Transfection Reagent (Life Technologies, Carlsbad, CA, U.S.A.) at 37°C. After incubation for 4 h, the culture medium was replaced with fresh complete medium containing CA or TA. Cells were cultured at 37°C for 3 d and then lysed in 100 µL Cell Culture Lysis Reagent (Promega Corp., Madison, WI, U.S.A.). Luciferase activity was measured using a Luminescencer-PSN luminometer (ATTO Co., Ltd., Tokyo, Japan) with 5 µL aliquots of the cell solution mixed with 10 µL luciferase assay substrate (PicaGene, Toyo Ink Mfg. Co., Ltd., Tokyo, Japan).
Evaluation of Egress Efficiency of HCV ParticlesOKH-4 cells were seeded at 5 × 104 cells per well in 48-well plates and cultured at 37°C for 1 d. HCV-JFH1 full genomic RNA (1 µg each/well) from pJFH1 was transfected into cells using DMRIE-C Transfection Reagent (Life Technologies, Carlsbad, CA, U.S.A.) at 37°C. After incubation for 4 h, the culture medium was replaced with fresh complete medium containing CA or TA. Cells were further incubated at 37°C for 3 d and intracellular and extracellular HCV-JFH1 RNA contents were determined by qRT-PCR as described above.
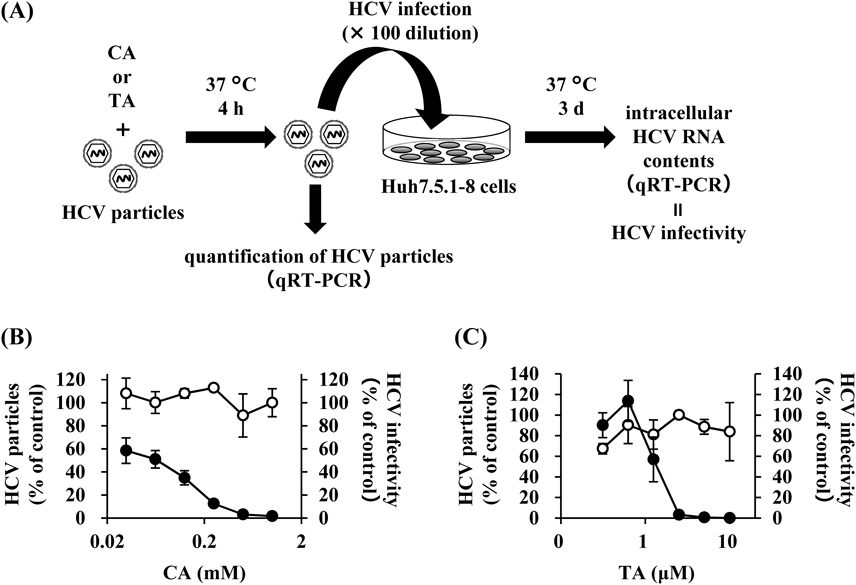
Evaluation of Infectivity of Viral ParticlesHuh7.5.1–8 cells were seeded at 5 × 104 cells in 48-well plate and cultured at 37°C for 1 d. HCV-JFH1-containing medium (corresponding to a multiplicity of infection (MOI) of 1.0; 200 µL) was mixed with various concentrations of CA or TA at 37°C for 4 h. Cells were then infected with a 100-fold dilution of these HCV-JFH1-containing media in order to reduce the direct influence of the compounds on the cells. At 3 d post-infection (p.i.), cellular HCV RNA copies were determined by qRT-PCR as described above.
HCV Particle Cellular Attachment AssayHuh7.5.1–8 cells were seeded at 5 × 104 cells per well in 48-well plates and cultured for 2 d. HCV-JFH1 (corresponding to an MOI of 10; 900 µL) were pretreated with CA (1 mM) or TA (10 µM) at 37°C for 4 h. Huh7.5.1–8 cells were then incubated with these HCV-JFH1 (200 µL/well) at 4°C for 2 h. After cells were washed thrice with PBS, amounts of HCV-JFH1 particles attached to the cells were quantified by qRT-PCR.
Evaluation of ApoE Associated with HCV Particles by Co-immunoprecipitation AssayHCV-JFH1-containing medium (corresponding to an MOI of 10; 200 µL) were pretreated with CA (1 mM) or TA (10 µM). Anti-ApoE pAb (5 µL) was then added to the medium. These mixtures (150 µL/aliquot) were treated with 50 µL of 50% (v/v) protein A/G PLUS agarose (Santa Cruz, Dallas, TX, U.S.A.) in PBS at 4°C for 2 h. Immunoprecipitated fractions were washed trice with PBS. Then, 150 µL 10% SDS was added to these tubes and heated at 70°C for 10 min. After centrifugation at 15 krpm (20400 × g) for 1 min, supernatants were collected and HCV-JFH1 RNA fractions were purified using FavorPrep Viral nucleic acid extraction kit I (FAVORGEN Biotech Corp.).
RESULTS
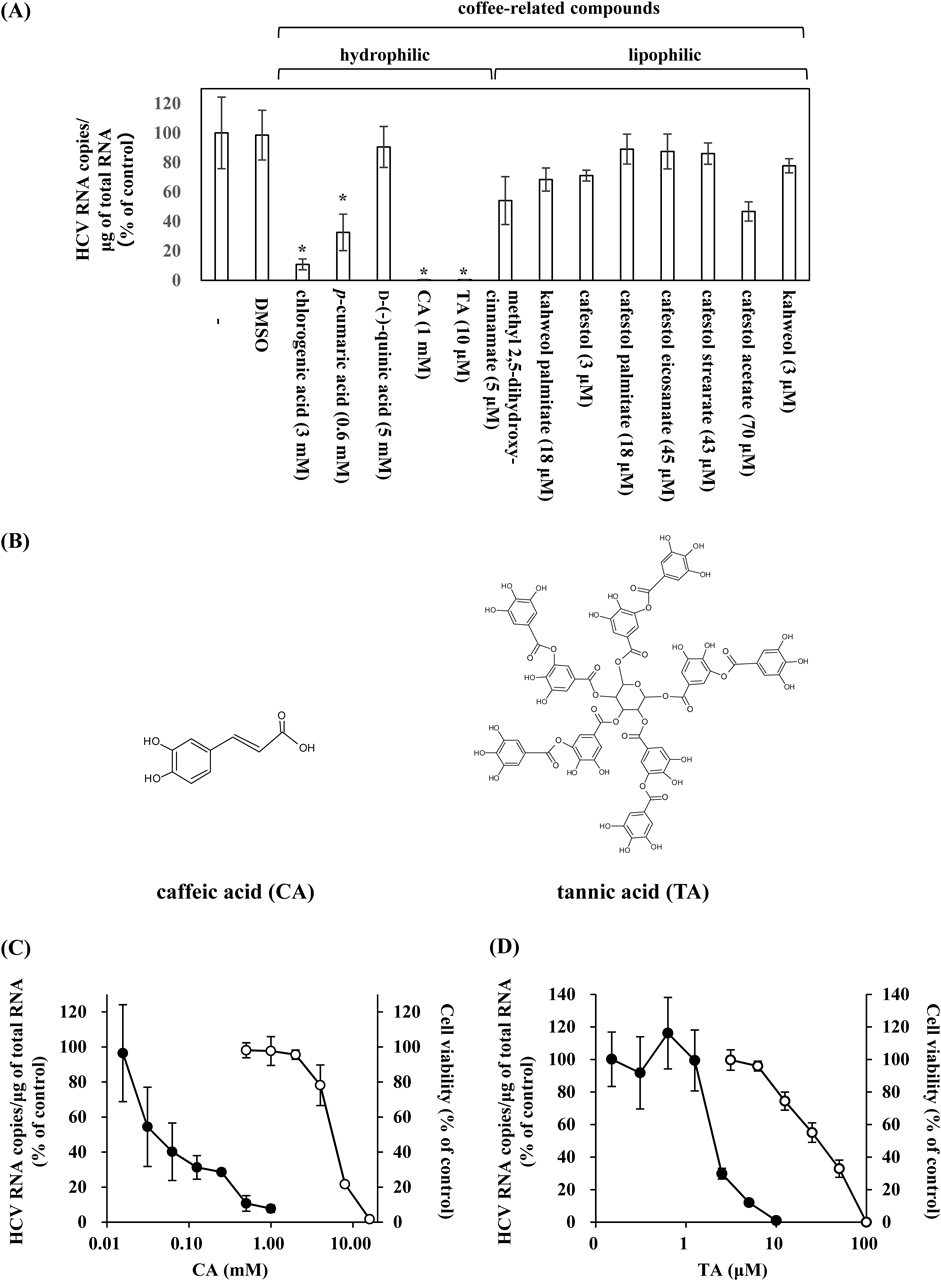
Inhibitory Effects of CA and TA on HCV InfectionPreviously, we showed that a coffee extract in addition to coffee-related compounds such as CA and p-coumaric acid can inhibit HCV infection.11) In the present study, we explored the effects of a variety of other coffee-related compounds, including the hydrophilic group (chlorogenic acid, p-coumaric acid, quinic acid, CA, and TA) and the lipophilic group (methyl 2,5-dihydroxy-cinnamate, kahweol palmitate, cafestol, cafestol palmitate, cafestol eicosanate, cafestol strearate, cafestol acetate, and kahweol), on HCV infection. Among the hydrophilic group, chlorogenic acid, p-cumaric acid, CA, and TA had apparent inhibitory effects on HCV infection, but no compounds in the lipophilic group displayed these effects (Fig. 1A). In particular, CA and TA (Fig. 1B) showed stronger inhibitory effects (Fig. 1A), and we examined whether these effects were dose-dependent and whether they resulted in cellular toxicity. We found that both CA (Fig. 1C) and TA (Fig. 1D) suppressed cellular HCV RNA levels in a dose-dependent manner. Based on immunoblot and immunofluorescence analyses, the cellular contents of viral proteins, HCV core and NS3, were also decreased in a dose-dependent manner by treatment with CA and TA (Supplementary Figs. 1A–C). CA and TA exhibited IC50 values of 0.06 ± 0.01 mM and 2.56 ± 1.32 µM and CC50 values of 2.40 ± 0.79 mM and 28.21 ± 7.63 µM, giving selectivity index estimates (SI) of 40 and 11, respectively (Table 1).
Table 1. Anti-HCV Activity and Cytotoxicity of CA and TA
| Compound | CC50 | IC50 | CC50/IC50 |
|---|
| Caffeic acid (CA) | 2.40 ± 0.79 mM | 0.06 ± 0.01 mM | 40 |
| Tannic acid (TA) | 28.21 ± 7.63 µM | 2.56 ± 1.32 µM | 11 |
Next, we determined which steps in the HCV lifecycle were affected by CA and TA. First, we tested the effects of CA and TA on HCV replication using Huh7.5.1–8 cells carrying a subgenomic replicon with the luciferase reporter gene, SGR-JFH1/Luc.26) Even at a concentration of 1 mM (Fig. 2A), at which HCV infection was strongly inhibited by CA (Fig. 1), luciferase activities were unaffected. Meanwhile, luciferase activities in the TA-treated cells were decreased in a dose-dependent manner and reached a plateau of 40–60% at 5 µM TA (Fig. 2B).
Next, we examined the effects of CA and TA on viral egress efficiency (Fig. 2C). OCLN-knockout OKH-4 cells, which were not permissive to HCV entry and thus did not support secondary infection, were transfected with full-length HCV RNA genomes and then treated with CA and TA. At 1–3 d post-transfection (p.t.), intracellular and extracellular HCV RNA was quantified by qRT-PCR. We calculated HCV egression activity as the ratio of extracellular/intracellular HCV RNA (Fig. 2C). HCV egression efficiency was not affected by treatment with CA, but was significantly reduced by treatment with TA (Fig. 2D).
Furthermore, we investigated the effects of CA and TA on infectious HCV particles themselves (Fig. 3A). HCV-JFH1 particles were treated with various concentrations of CA or TA at 37°C for 4 h and then inoculated into Huh7.5.1–8 cells. We then evaluated the infectivity of the HCV particles by measuring the HCV RNA contents in the inocula and the cellular HCV RNA levels at 3 d p.i. Neither CA nor TA affected the amounts of HCV-JFH1 in the inocula, but both decreased the HCV RNA levels in the infected Huh7.5.1–8 cells (Figs. 3B, C). CA and TA exhibited IC50 values of 0.05 ± 0.01 mM and 1.31 ± 0.89 µM, respectively, similar to the values in Table 1. Cellular HCV core proteins were also decreased in a dose-dependent manner by treatment of HCV-JFH1 particles with CA and TA (Supplementary Figs. 2A, B). Furthermore, CA and TA pretreatment of HCV particles decreased the infectivity of other infectious HCV particles, such as HCV-TNS2J1 (genotype 1b/2a chimera) and HCV-Jc1 (genotype 2a/2a chimera), in a dose-dependent manner (Supplementary Figs. 3A–D).
CA-Treated HCV Particles Showed Lower Cellular AttachmentSince CA and TA attenuated the infectivity of HCV particles, we investigated whether CA and TA affect the cellular attachment of HCV particles. HCV-JFH1 were pretreated with CA (1 mM) or TA (10 µM) at 37°C for 4 h. Huh7.5.1–8 cells were then incubated with these HCV particles at 4°C for 2 h. After cells were washed thrice with PBS, the amount of HCV-JFH1 particles attached to the cells was quantified by qRT-PCR. There was no change in cellular HCV RNA levels after treatment with TA relative to the control, treated with dimethyl sulfoxide (DMSO). On the other hand, CA treatment significantly reduced cellular HCV RNA levels by approximately 25% (Fig. 4).
CA Prevented the Interaction Between HCV Particles and ApoEIt is well known that the formation of a complex between HCV particles and ApoE is critical for the infectivity of HCV particles.27–32) Therefore, we explored the possibility that CA interrupts the interaction between HCV particles and ApoE. The amounts of HCV particles bound to ApoE in the presence or absence of CA or TA were determined by immunoprecipitation assays using anti-ApoE antibody (Fig. 5A).
Consistent with the results of a previous study,33) 70–80% of HCV particles (as viral RNA contents) were immunoprecipitated with anti-ApoE antibody when non-treated or DMSO-treated HCV particles were analyzed (Fig. 5B). A similar result was achieved with TA-treated HCV particles (Fig. 5B), indicating that TA had no effects on the HCV-ApoE interaction. NP40-treated HCV particles, which have no envelopes, were used as a negative control; approximately 24% were immunoprecipitated with anti-ApoE antibody (Fig. 5B); likewise, approximately 29% of CA-treated HCV particles were immunoprecipitated with anti-ApoE antibody.
DISCUSSION
In the present study, we found that TA, in addition to CA,11) strongly prevented HCV-JFH1 infection in the cell culture system (Fig. 1, Supplementary Fig. 1, Table 1). TA affected the whole HCV lifecycle including viral entry, genome replication, and egress (Figs. 2B, D, 3C, Supplementary Figs. 2B, 3C, D). Although further analyses are needed to understand the molecular mechanism underlying these effects, TA mainly acts on viral particles (Fig. 3) and inhibits the late entry step after the cellular attachment of viral particles (Figs. 4, 5). On the other hand, CA showed no effect on the genome replication or egress processes of HCV-JFH1 infection (Figs. 2A, D), but did inhibit viral entry, especially at the early stage before viral attachment to the host cells (Fig. 3B). These effects were also confirmed in other viral strains, including Jc1 and TNS2J1 (Supplementary Fig. 3), suggesting that CA and TA have general inhibitory effects on HCV infection.
It is well known that formation of a complex of HCV particles and ApoE is critical for viral infectivity.27–29) These complexes, named lipoviroparticles (LVPs), enhance the interaction between HCV particles and cellular receptors, including heparan sulfate proteoglycans34) such as syndecan 4,35) low density lipoprotein (LDL) receptors,36) and scavenger receptors class B type I,37) resulting in efficient viral attachment to and entry into host cells.28,38) We demonstrated that CA interrupted the association between ApoE and HCV particles (Fig. 5B) and reduced the cellular attachment of viral particles (Fig. 4), and we therefore concluded that this is the major molecular mechanism underlying the inhibition of HCV infection by CA.
Recently, it was reported that CA prevented HCV genome replication,13,14) but we did not observe this effect in the present study (Fig. 2). This might be explained by differences in researchers’ use of host cells and viral/replicon strains. In the present study, we used Huh7.5.1–8 cells and HCV-JFH1 (genotype 2a), while previous studies have used Huh7,13,14) Huh7.5.1,13) Con1 replicon (genotype 1b), J399EM (genotype 2a), a JFH-1-based adaptive strain in which an enhanced green fluorescent protein (EGFP) gene was inserted into NS5A,13) and N replicon (genotype 1b).15)
It has also been reported that CA has inhibitory effects on other viruses such as hepatitis B virus (HBV), canine distemper virus (CDV), herpes simplex virus type 1 (HSV-1), influenza A virus (IAV), polio virus type 1 (PV-1), and severe fever with thrombocytopenia syndrome virus (SFTSV) in cultured cells.12,14–17,19,21,39) CA has also shown inhibitory activity against human immunodeficiency virus (HIV) integrase (IC50 = 2.8 µM),20,40) which is a critical enzyme for HIV replication. HBV infection was prevented by CA mainly at the viral genome replication step.12) CDV infection was inhibited by CA treatments at 1 and 2 h p.i., but not at −1 and 0 h p.i.,39) suggesting that CA affects CDV after entry into cells. It is thought that CA primarily affects the early stages of HSV and IAV infection, although its target has not yet been clearly identified.15,16) Unlike in HCV in the present study, CA has not been shown to have virucidal effects on HSV-1 or IAV.15,16) In PV-1 infection, CA treatment of cells during infection resulted in higher viral inhibition than CA treatment at pre- or post-infection.17) These results could be partially explained if one assumes that CA affects PV-1 particles and thus blocks viral entry, as with HCV infection, although further detailed investigations are needed. Very recently, we reported that CA directly acts on SFTSV particles and reduces their infectivity.21) The effect of CA on SFTSV is very similar to that on HCV shown in the present study, suggesting cognate underlying molecular mechanisms.
In the present study, we found that the coffee-related compounds CA and TA act on HCV particles and abrogate their infectivity. Although further investigation, including in vivo experiments, is necessary, intake of coffee or the coffee-related compounds CA and TA may lead to prevention of HCV infection and slower disease progression after HCV infection. Foods and food ingredients such as CA and TA, which are inexpensive and easy to supply, should be considered in global efforts to control HCV-derived diseases.
Acknowledgments
We thank all members of our laboratory, especially Drs. Kyoko Saito and Yoshimi Shimizu, for their useful comments. This study was supported by a Research Grant from the All Japan Coffee Association and partly by the Research Program on Hepatitis from the Japan Agency for Medical Research and Development (AMED) (Grant No. 17fk0210308j0003 to M.F.), AMED-CREST (Grant No. JP18gm0910005j0004 to K.H.) and JSPS KAKENHI (Grant Nos. 16K08260 and 18H02856 to M.F.).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, Netesov S, Nishioka K, Shin-i T, Simmonds P, Smith D, Stuyver L, Weiner A. International Committee on Virus Taxonomy. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol., 143, 2493–2503 (1998).
- 2) World Health Organization. “2017. Hepatitis C fact sheet.”: ‹www.who.int/mediacentre/factsheets/fs164/en/›, Accessed 16 May 2018.
- 3) Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol., 61 (Suppl.), S79–S90 (2014).
- 4) El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology, 142, 1264–1273.e1 (2012).
- 5) Spengler U. Direct antiviral agents (DAAs)—A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther., 183, 118–126 (2018).
- 6) Braillon A. Interferon-free treatments against HCV are far from free. Lancet, 386, 856 (2015).
- 7) Machado MM, Montagner GFFdS, Aline Boligon MLA, Rocha MIUMd, Lera JPB, Cruz CBeIBMd. Determination of polyphenol contents and antioxidant capacity of no-alcoholic red grape products (vitis labrusca) from conventional and organic crops. Quim. Nova, 34, 798–803 (2011).
- 8) Zheng W, Wang SY. Oxygen radical absorbing capacity of phenolics in blueberries, cranberries, chokeberries, and lingonberries. J. Agric. Food Chem., 51, 502–509 (2003).
- 9) Chang H, Wang Y, Yin X, Liu X, Xuan H. Ethanol extract of propolis and its constituent caffeic acid phenethyl ester inhibit breast cancer cells proliferation in inflammatory microenvironment by inhibiting TLR4 signal pathway and inducing apoptosis and autophagy. BMC Complement. Altern. Med., 17, 471 (2017).
- 10) Duan J, Xiaokaiti Y, Fan S, Pan Y, Li X, Li X. Direct interaction between caffeic acid phenethyl ester and human neutrophil elastase inhibits the growth and migration of PANC-1 cells. Oncol. Rep., 37, 3019–3025 (2017).
- 11) Tanida I, Shirasago Y, Suzuki R, Abe R, Wakita T, Hanada K, Fukasawa M. Inhibitory effects of caffeic acid, a coffee-related organic acid, on the propagation of hepatitis C virus. Jpn. J. Infect. Dis., 68, 268–275 (2015).
- 12) Wang GF, Shi LP, Ren YD, Liu QF, Liu HF, Zhang RJ, Li Z, Zhu FH, He PL, Tang W, Tao PZ, Li C, Zhao WM, Zuo JP. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antiviral Res., 83, 186–190 (2009).
- 13) Shen J, Wang G, Zuo J. Caffeic acid inhibits HCV replication via induction of IFNα antiviral response through p62-mediated Keap1/Nrf2 signaling pathway. Antiviral Res., 154, 166–173 (2018).
- 14) Shen H, Yamashita A, Nakakoshi M, Yokoe H, Sudo M, Kasai H, Tanaka T, Fujimoto Y, Ikeda M, Kato N, Sakamoto N, Shindo H, Maekawa S, Enomoto N, Tsubuki M, Moriishi K. Inhibitory effects of caffeic acid phenethyl ester derivatives on replication of hepatitis C virus. PLOS ONE, 8, e82299 (2013).
- 15) Ikeda K, Tsujimoto K, Uozaki M, Nishide M, Suzuki Y, Koyama AH, Yamasaki H. Inhibition of multiplication of herpes simplex virus by caffeic acid. Int. J. Mol. Med., 28, 595–598 (2011).
- 16) Utsunomiya H, Ichinose M, Ikeda K, Uozaki M, Morishita J, Kuwahara T, Koyama AH, Yamasaki H. Inhibition by caffeic acid of the influenza A virus multiplication in vitro. Int. J. Mol. Med., 34, 1020–1024 (2014).
- 17) Búfalo MC, Figueiredo AS, de Sousa JP, Candeias JM, Bastos JK, Sforcin JM. Anti-poliovirus activity of Baccharis dracunculifolia and propolis by cell viability determination and real-time PCR. J. Appl. Microbiol., 107, 1669–1680 (2009).
- 18) Özçelik B, Kartal M, Orhan I. Cytotoxicity, antiviral and antimicrobial activities of alkaloids, flavonoids, and phenolic acids. Pharm. Biol., 49, 396–402 (2011).
- 19) Arakawa T, Yamasaki H, Ikeda K, Ejima D, Naito T, Koyama AH. Antiviral and virucidal activities of natural products. Curr. Med. Chem., 16, 2485–2497 (2009).
- 20) Bailly F, Cotelle P. Anti-HIV activities of natural antioxidant caffeic acid derivatives: toward an antiviral supplementation diet. Curr. Med. Chem., 12, 1811–1818 (2005).
- 21) Ogawa M, Shirasago Y, Ando S, Shimojima M, Saijo M, Fukasawa M. Caffeic acid, a coffee-related organic acid, inhibits infection by severe fever with thrombocytopenia syndrome virus in vitro. J. Infect. Chemother., 24, 597–601 (2018).
- 22) Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A., 102, 9294–9299 (2005).
- 23) Shirasago Y, Sekizuka T, Saito K, Suzuki T, Wakita T, Hanada K, Kuroda M, Abe R, Fukasawa M. Isolation and characterization of an Huh.7.5.1-derived cell clone highly permissive to hepatitis C virus. Jpn. J. Infect. Dis., 68, 81–88 (2015).
- 24) Shirasago Y, Shimizu Y, Tanida I, Suzuki T, Suzuki R, Sugiyama K, Wakita T, Hanada K, Yagi K, Kondoh M, Fukasawa M. Occludin-knockout human hepatic Huh7.5.1–8-derived cells are completely resistant to hepatitis C virus infection. Biol. Pharm. Bull., 39, 839–848 (2016).
- 25) Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med., 11, 791–796 (2005).
- 26) Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J. Clin. Microbiol., 43, 5679–5684 (2005).
- 27) Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol., 81, 13783–13793 (2007).
- 28) Jiang J, Wu X, Tang H, Luo G. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLOS ONE, 8, e67982 (2013).
- 29) Fukuhara T, Wada M, Nakamura S, Ono C, Shiokawa M, Yamamoto S, Motomura T, Okamoto T, Okuzaki D, Yamamoto M, Saito I, Wakita T, Koike K, Matsuura Y. Amphipathic alpha-helices in apolipoproteins are crucial to the formation of infectious hepatitis C virus particles. PLOS Pathog., 10, e1004534 (2014).
- 30) Li Z, Li Y, Bi Y, Zhang H, Yao Y, Li Q, Cun W, Dong S. Extracellular interactions between hepatitis C virus and secreted apolipoprotein E. J. Virol., 91, e02227-16 (2017).
- 31) Bankwitz D, Doepke M, Hueging K, Weller R, Bruening J, Behrendt P, Lee JY, Vondran FWR, Manns MP, Bartenschlager R, Pietschmann T. Maturation of secreted HCV particles by incorporation of secreted ApoE protects from antibodies by enhancing infectivity. J. Hepatol., 67, 480–489 (2017).
- 32) Roingeard P, Dreneau J, Meunier JC. Unravelling the multiple roles of apolipoprotein E in the hepatitis C virus life cycle. Gut, 66, 759–761 (2017).
- 33) Benedicto I, Gondar V, Molina-Jimenez F, Garcia-Buey L, Lopez-Cabrera M, Gastaminza P, Majano PL. Clathrin mediates infectious hepatitis C virus particle egress. J. Virol., 89, 4180–4190 (2015).
- 34) Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol., 86, 7256–7267 (2012).
- 35) Lefèvre M, Felmlee DJ, Parnot M, Baumert TF, Schuster C. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLOS ONE, 9, e95550 (2014).
- 36) Owen DM, Huang H, Ye J, Gale M Jr. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology, 394, 99–108 (2009).
- 37) Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K. Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J. Virol., 84, 12048–12057 (2010).
- 38) Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology, 56, 484–491 (2012).
- 39) Wu ZM, Yu ZJ, Cui ZQ, Peng LY, Li HR, Zhang CL, Shen HQ, Yi PF, Fu BD. In vitro antiviral efficacy of caffeic acid against canine distemper virus. Microb. Pathog., 110, 240–244 (2017).
- 40) Singh SB, Jayasuriya H, Dewey R, Polishook JD, Dombrowski AW, Zink DL, Guan Z, Collado J, Platas G, Pelaez F, Felock PJ, Hazuda DJ. Isolation, structure, and HIV-1-integrase inhibitory activity of structurally diverse fungal metabolites. J. Ind. Microbiol. Biotechnol., 30, 721–731 (2003).