Current Topics: Reviews
Transcription Factors and Downstream Genes in Cadmium Toxicity
2019 年 42 巻 7 号 p. 1083-1088
詳細
2019 年 42 巻 7 号 p. 1083-1088
Cadmium (Cd) is a harmful heavy metal widely present in the environment which can cause severe kidney damage. The proximal tubular cells are the main target of renal Cd toxicity. The consequences of Cd cytotoxicity involve apoptosis and necrosis. Recently, we and others have focused on how Cd affects transcription factors and the regulation of their target genes. Those studies showed that transcription factors initiate numerous pathways upon Cd exposure, leading to apoptosis, autophagic cell death, disruption of cell–cell adhesion, and generation of mitochondrial reactive oxygen species. Of particular note, Cd induces endoplasmic reticulum stress, resulting in not only apoptosis but also autophagic dysregulation, which can trigger cell damage. In some cases, however, Cd-regulated transcription factors can induce cell survival signaling. This review centers on our own research to elucidate the transcription factor-downstream gene cascades that are central to Cd-induced renal toxicity.
Cadmium (Cd) is a heavy metal that is widespread in the environment. Cd causes itai-itai disease, the main symptom of which is renal disorder and accompanying osteomalacia.1) Chronic Cd exposure via polluted water or food, or cigarettes, induces nephrotoxicity, especially damage of proximal tubular cells.2–4) The biological half-life of Cd in humans varies with gender, age, and exposure level but is approximately 10–30 years.3,4) Therefore, the amount of Cd accumulated in the body increases with age, as do the health effects associated with Cd accumulation.5)
Over the past 15 to 20 years, the relative roles of apoptosis, necrosis, and the autophagic process have been proposed for Cd-induced cytotoxicity. In the 1990 s, several studies suggested that the early phase of Cd cytotoxicity involved the induction of apoptotic signals.6–8) In the 2000 s, Cd was reported to release cytochrome c and apoptosis-inducing factor (AIF) from mitochondria. The released cytochrome c and AIF induce apoptosis caspases dependently or independently.9–11) Cd also upregulates kidney injury molecule 1 (Kim-1) expression before necrosis, but with a modest level of apoptosis in rat proximal tubules.12) In renal tubular cells, Cd can also elevate cytosolic calcium levels via release from the endoplasmic reticulum (ER), an intracellular calcium reservoir. Elevated cytosolic calcium causes apoptosis through the calpain-caspase pathway.13,14) Chargui et al. demonstrated the activation of autophagy as the repair process in the absence of an apoptosis signal in rat proximal tubules.15)
Although several pathways involved in Cd-induced cell death have been reported, the factors initiating these pathways remain to be fully elucidated. To clarify the molecular mechanism that initiates Cd-induced nephrotoxicity, toxicogenomic methods to search for molecules involved in Cd renal toxicity have recently been applied by ourselves and other groups. In particular, using protein/DNA-binding arrays and small interfering RNA (siRNA) tools, we searched for candidate transcription factors that regulate the expression of genes involved in Cd toxicity. Other, diverse studies have also shown relationships between transcription factors and target genes responsible for the initiation of Cd renal toxicity.
This review describes transcription factors that regulate the expression of genes that induce toxic effects, such as apoptosis, autophagy, disruption of cell–cell adhesion, and mitochondrial reactive oxygen species (ROS) generation.
Several studies have indicated transcription factors that downregulate genes related to Cd-induced apoptosis in the kidney. Some representative findings are described below.
2.1. Cd Decreases Inhibitor of Apoptosis Protein 1/2 Levels via Suppression of Nuclear Factor-Kappa B ActivityNuclear factor-kappa B (NF-κB) is a well-known transcription factor that plays a key role in modulating cell death or survival. Xie and Shaikh revealed that Cd suppresses the DNA binding activity of NF-κB and levels of inhibitor of apoptosis protein 1 (IAP1) and IAP2 (also known as BIRC2 and BIRC3) in rat proximal tubular NRK-52E cells.16) IAP1 and IAP2 are target genes of NF-κB, and IAP1 and IAP2 bind to caspases, which are key inducers of apoptosis.17) NF-κB induces IAP1 and IAP2 to inhibit apoptosis. Therefore, Cd increases the levels of caspases 3, 7, and 9 and induces caspase-dependent apoptosis via suppression of NF-κB16) (Fig. 1).

Cd induces caspase-dependent apoptosis via inactivation of NF-κB and downregulation of IAPs. Furthermore, Cd-induced ER stress triggers CHOP- or JNK-dependent apoptosis via activation of ATF4, ATF6, and XBP1.
Cd contributes to ER stress, and the eukaryotic initiation factor 2α-subunit (eIF2α)-activating transcription factor 4 (ATF4) pathway is involved in cell adaptation to stress.18) ER stress phosphorylates eIF2α and translocates ATF4 to the nucleus from the cytoplasm. Komoike et al. demonstrated that apoptosis is induced by Cd following phosphorylation of eIF2α and activation of ATF4 in human renal proximal tubular HK-2 cells19) (Fig. 1). Cd increased CCA AT/enhancer-binding protein-homologous protein (CHOP) and downregulated ATF4. The generation of CHOP is responsible for the induction of apoptosis.
Yokouchi et al. showed that Cd induces ER stress in a porcine renal proximal tubular cell line, LLC-PK1.20,21) It is well known that activating transcription factor 6 (ATF6) and inositol-requiring ER-to-nucleus signal kinase 1 (IRE1) are two of three major transmembrane transducers for sensing the ER. ATF6 regulates the expression of CHOP and X-box binding protein 1 (XBP1) genes.22) XBP1 mRNA is spliced by IRE1, and XBP1 is able to act as a transcription factor.23) The induction of CHOP by ATF6 and IRE1-initiated activation of XBP1 are responsible for the induction of apoptosis. Cd activates ATF6 and elevates CHOP expression.20,21) On the other hand, Cd upregulates IRE1 to activate XBP1. Furthermore, IRE1-c-Jun N-terminal kinase (JNK) is a known proapoptotic signaling pathway,24) and Cd has been shown to phosphorylate JNK. Yokouchi et al. proposed the existence of the XBP1-JNK proapoptotic pathway involved in ER stress-induced apoptosis20,21) (Fig. 1).
2.3. Cd Downregulates UBE2D Family Gene Expression via Suppression of YY1 and FOXF1We reported that Cd inhibits the gene expression of the Ube2d family (Ube2d1, Ube2d2, Ube2d3, and Ube2d4) prior to toxic effects in NRK-52E cells and in the kidney of C57BL/6J mice.25,26) The Ube2d family consists of ubiquitin (Ub)-conjugating enzymes (E2), which tag substrate proteins for degradation with Ub. Ube2d ubiquitinates the apoptosis-inducing factor p53, which is then degraded by the proteasome. Because of a lack of Ub conjugation, p53 is accumulated in the cells in the presence of Cd.26,27) In HK-2 cells, among the UBE2D family, Cd only inhibited the expression of UBE2D2 and UBE2D4.27) Lee et al. showed that knockdown of both UBE2D2 and UBE2D4 resulted in the accumulation of p53 equivalent to Cd-induced p53.27) Cd increased the level of phosphorylated p53 in the nuclei of HK-2 cells, and knockdown of p53 prevented Cd-induced apoptosis. Moreover, apoptotic cells were positive for p53 in the proximal tubules of mice administered Cd for 6 months. These results show that Cd-induced accumulation of p53 and Cd-induced p53-dependent apoptosis result from the inhibition of UBE2D family gene expression.
Protein/DNA arrays were used to identify transcription factors of which the activities are altered by nontoxic levels of Cd in NRK-52E and HK-2 cells.27–29) Protein/DNA arrays screen a maximum of 345 transcription factors with activities that can be affected by a test substance. Cd downregulated the activity of a diverse set of transcription factors. Among these, we focused on yin yang 1 (YY1) and forkhead box F1 (FOXF1) because they have target binding sequences upstream of both UBE2D2 and UBE2D4.27) Cd inhibited mRNA and protein levels of FOXF1, but those of YY1 were not changed. Knockdown of YY1 downregulated the gene expression of UBE2D2 but not UBE2D4. However, knockdown of FOXF1 downregulated the gene expression of UBE2D4 but not UBE2D2. Therefore, Cd decreased the gene expression of UBE2D2 and UBE2D4 following inhibition of YY1 and FOXF1, respectively (Fig. 2).
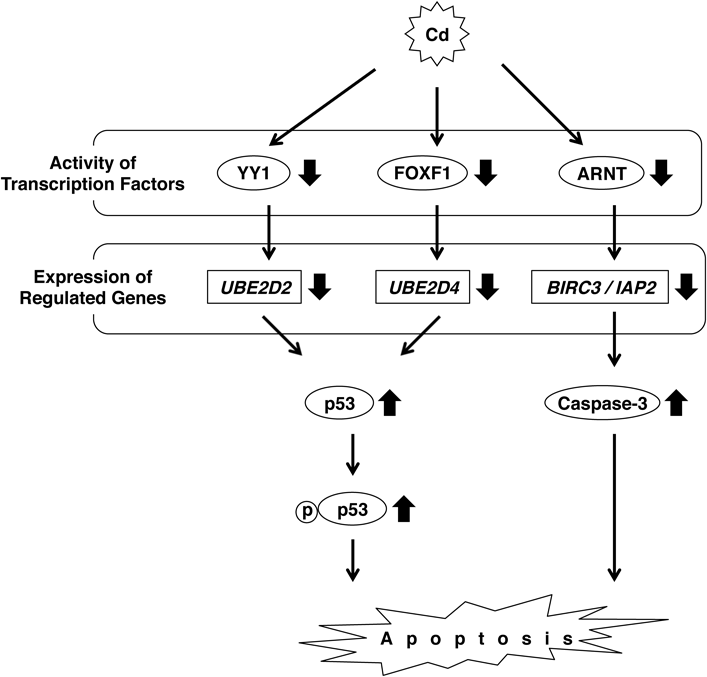
Cd induces p53-dependent apoptosis via downregulation of UBE2D2 and UBE2D4, causing inactivation of YY1 and FOXF1, respectively. Furthermore, Cd induces caspase-3-dependent apoptosis via inactivation of ARNT and decreased BIRC3 expression.
We discovered another route for Cd induction of renal toxicity. The protein/DNA arrays described above showed that Cd decreased the activity of the transcription factor aryl hydrocarbon receptor nuclear translocator (ARNT) in HK-2 cells. Knockdown of ARNT decreased cell viability and inhibited the expression of 27 genes, indicating that these genes are downstream of ARNT. Among these 27 genes, baculoviral IAP repeat-containing protein 3 (BIRC3) was markedly inhibited by Cd.29) BIRC3 is a member of the BIRC family (BIRC1–8) also known as the IAP family, i.e., BIRC1/NAIP, BIRC2/cIAP1, BIRC3/cIAP2, BIRC4/XIAP, BIRC5/Survivin, BIRC6/Apollon, BIRC7/ML-IAP, and BIRC8/ILP.30,31) BIRC3 inhibits apoptosis by interfering with caspase activation.32) Cd increased the level of cleaved caspase-3, the active form of caspase-3 in HK-2 cells. Similarly, knockdown of BIRC3 activated caspase-3 and increased cytotoxicity following the induction of apoptosis (Fig. 2).
Cd-induced renal toxicity can be mediated by downregulated gene expression that affects processes other than apoptosis. Several representative findings are described below.
3.1. Cd Induces Cyclooxygenase 2-Mediated Autophagy via the eIF2α-ATF4 Pathway in ER StressCyclooxygenase 2 (COX2), the primary enzyme for the production of prostaglandin in the inflammatory reaction, is a downstream factor of ATF4.33) Luo et al. reported that Cd activates the eIF2α-ATF4 pathway in ER stress and induces COX2-dependent autophagy in human embryonic kidney (HEK) cells and in the mouse kidney34) (Fig. 3). Autophagy can be activated via inhibition of the phosphoinositide-3-kinase (PI3K)-Akt (serine/threonine protein kinase)-mammalian target of rapamycin (mTOR) pathway.35) COX2 knockdown increased levels of phosphorylated mTOR, leading to decreased autophagy and phosphorylated Akt.34)

Cd induces autophagy and mROS production and decreases cell–cell adhesion. Cd activates ATF4 and increases COX2 expression. Autophagy induced by Cd involves the inhibition of mTOR by COX2. Disruption of cell–cell adhesion triggered by Cd depends on the suppression of E-cadherin via activation of Snail. Cd produces mROS via inactivation of Foxo3a and decreases PGC1 and SOD2 expression.
E-Cadherin is responsible for cell–cell adhesion and is important for epithelial tissues to maintain their integrity.36) The zinc finger transcription factor Snail is a suppressor of E-cadherin and is activated by Notch1.37) The Notch pathway is highly conserved and is widely involved in tissue patterning and morphogenesis, cell differentiation, proliferation, and death.38) In addition, Notch1 is controlled by the PI3K/Akt pathway. Fujiki et al. demonstrated that Cd increased Notch1 and Snail levels via phosphorylation of PI3K and Akt and disrupted E-cadherin-mediated cell–cell adhesion in HK-2 cells39) (Fig. 3).
3.3. Cd Induces Mitochondrial ROS via Suppression of Sirt3 and FoxO3aSirtuin 3 (Sirt3) is the primary mitochondrial acetyl-lysine deacetylase that modulates various proteins to control mitochondrial function.40) Sirt3 directly binds and deacetylates superoxide dismutase 2 (SOD2), which increases SOD2 activity and maintains mitochondrial ROS (mROS) homeostasis.41,42) In human hepatocellular carcinoma HepG2 cells and mouse liver, Cd induces hepatotoxicity via mitochondrial-derived superoxide anion-dependent autophagy through the Sirt3-SOD2 pathway.43)
In addition to the action of Sirt3 in hepatocytes, Fu et al. confirmed the Sirt3-SOD2 pathway as a mechanism to inhibit Cd renal toxicity.44) Mitochondrial Sirt3 induces forkhead box O3 (FoxO3a) translocation to the nucleus and upregulation of peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α) and SOD2. Phosphorylation of FoxO3a inactivates FoxO3a. PGC-1α and SOD2 suppress ROS production and protect cells from mROS-induced oxidative damage.45) Fu et al. showed that Cd suppresses FoxO3a activity (the level of phosphorylated FoxO3a was increased) and expression of PGC-1α and SOD2.44) Therefore, Cd induces mROS via inhibition of the Sirt3-FoxO3a pathway in mouse renal tubular epithelial TCMK-1 cells (Fig. 3).
3.4. Cell Protection against Cd via Regulation of ATF4 and FoxO3aFujiki et al. demonstrated that increased ATF4 expression induced through the PI3K/Akt pathway plays a role in the survival of HK-2 cells.46) Activated ATF4 and ATF6 induced not only CHOP but also the 78-kDa glucose-regulated protein (GRP78), an ER-resident molecular chaperone, in Cd-treated HK-2cells.19–21) Luo et al. also showed that Cd induced COX2 and GRP78 in vivo and in vitro via ATF4.34) GRP78 is an antiapoptosis molecule that protects against Cd-induced renal toxicity. Furthermore, FOXO3a is phosphorylated by the PI3K/Akt pathway, which is protective for Cd-treated HK-2 cells.47)
Cd affects diverse biological processes and causes renal toxicity through the induction of apoptosis and autophagy, generation of mROS, and disruption of cell–cell adhesion. These processes are complicated, and thus the mechanism of Cd-induced toxicity has not been entirely clarified. Although various studies reported individual biological factors affected by Cd, few indicated the complete process from upstream factors (e.g., transcription factors, enzymes) to the final stage of toxic manifestation. This review presents targets of Cd renal toxicity and associated molecular mechanisms, especially for transcription factors and their downstream genes.
Cd-induced apoptosis involves numerous pathways (Figs. 1, 2). Cd inactivates NF-κB and induces caspase-mediated apoptosis via downregulation of IAP1 and IAP2.16) The ARNT-BIRC3 pathway also induces caspase-mediated apoptosis29) (Fig. 2). The expression of BIRC3 was specifically decreased, although that of BIRC4 was increased, and the expression of other BIRC family members was unaffected by Cd treatment in HK-2 cells.29) These results indicate that Cd-induced caspase-dependent apoptosis is followed by the suppression of each IAP via activation of the respective transcription factors. Similarly, Cd decreases the gene expression of UBE2D2 and UBE2D4 in HK-2 cells via inactivation of YY1 and FOXF1, respectively27) (Fig. 2). Knockdown of YY1 does not affect UBE2D4 mRNA levels, and knockdown of FOXF1 does not decrease UBE2D2 mRNA levels.27) These findings indicate the specific relationship between a transcription factor and its regulated downstream gene. Meanwhile, both ATF4 and ATF6 upregulate CHOP in renal proximal tubular cells treated with Cd19,21) (Fig. 1). These results indicate that several transcription factors are involved in the expression of a single gene.
The upregulation of ATF4 induced COX2-dependent autophagy in Cd-treated HEK cells34) (Fig. 3). Similarly, inactivation of FoxO3a led to PGC1- and SOD2-mediated mROS production44) (Fig. 3). However, ATF4, ATF6, and FoxO3a can be associated with cell survival with Cd toxicity. It is suspected that ATF4, ATF6, and FoxO3a operate in a variety of signaling pathways and that Cd exposure induces numerous opposing effects simultaneously. Which effect predominates may depend on the dose and time of Cd exposure.
Recently, the expression of several novel genes has been suggested to change in Cd renal toxicity.48–50) Moreover, gestational exposure to Cd may affect fetal growth.51,52) Furthermore, metabolomic analysis indicated that several amino acid derivatives in mouse urine may be useful indicators of renal damage caused by long-term Cd exposure.53) These reports raise the expectation of finding unknown factors involved in Cd renal toxicity. As this review demonstrates, Cd leads to toxic effects through cascades of transcription factors and their regulated genes. To elucidate fully the renal toxicity caused by Cd, further research is required.
This work was partly supported by the Study of the Health Effects of Heavy Metals, organized by the Ministry of the Environment, Japan.
The authors declare no conflict of interest.