Current Topics: Reviews
Steroid-Resistant Asthma and Neutrophils
2020 Volume 43 Issue 1 Pages 31-35
Details
2020 Volume 43 Issue 1 Pages 31-35
Asthma patients are classified by phenotype and endotype. Although symptoms in most asthma patients are well controlled by glucocorticoid treatment, certain populations of severe eosinophilic asthma patients in T-helper 2 (Th2)/type 2 asthma and neutrophilic asthma patients in non-Th2/type 2 asthma show insensitivity to inhaled or oral glucocorticoid therapy. In some cases of severe eosinophilic asthma, eosinophils remain in the lungs despite glucocorticoid therapy. It was reported that interleukin (IL)-33-induced activation of type 2 innate lymphoid cells (ILC2) was resistant to glucocorticoid treatment in certain allergic conditions. Regarding neutrophilic airway inflammation in steroid-resistant asthma, IL-17 derived from Th17 cells and IL-8 and tumor necrosis factor-α derived mainly from macrophages were reported to be involved in the pathogenesis. Recently, “NETosis,” a specific cell death of neutrophils, has been reported to be involved in asthmatic airway inflammation. When NETosis is induced in asthma, aggravation of inflammation and delay of tissue repair could occur, suggesting that NETosis may be associated with the development of steroid-resistant asthma. This article reviews the pathogenesis of steroid-resistant asthma by focusing mainly on neutrophils.
Asthma is a chronic inflammatory airway disease characterized by airway hyperresponsiveness, remodeling, and eosinophilia. Long-term medication is required to control asthma pathogenesis. Inhaled glucocorticoids and long-acting β2 agonists have been used to suppress airway inflammation and bronchoconstriction, respectively, over the long term. The majority of asthma patients who respond well to inhaled glucocorticoid therapy are referred to as having “steroid-sensitive asthma.” However, in a certain population of asthma patients, symptoms cannot be well controlled by inhaled glucocorticoid or even by oral glucocorticoid therapy and their disease is referred to as “steroid-resistant asthma.”1,2) It has been suggested that 5–10% of asthma patients are steroid resistant.3)
Asthma is a heterogeneous disease, so that it exhibits marked variability in clinical and pathological features, indicating that there are different asthma phenotypes and endotypes. Wenzel1) proposed that asthma is classified as “T-helper 2 (Th2)/type 2 asthma” and “non-Th2/type 2 asthma” based on the inflammatory pattern. Furthermore, as shown in Table 1, it was proposed that Th2/type 2 asthma includes 1) early-onset allergic asthma, 2) late-onset eosinophilic asthma, and 3) exercise-induced asthma, and that non-Th2/type 2 asthma includes 4) obesity-related asthma and 5) neutrophilic asthma.1,2)
| Pathobiology | Steroid resistance | |
|---|---|---|
| Th2/type 2 inflammation | ||
| Early-onset allergic | Antigen-specific IgE, Th2 | |
| Late-onset eosinophilic | IL-5, eosinophilia | + |
| Exercise-induced | Mast cell, Th2, CysLTs | |
| Non-Th2/type 2 inflammation | ||
| Obesity-related | Lack of Th2, oxidative stress | + |
| Neutrophilic | Neutrophilia, Th17, IL-8 | + |
CysLTs, cysteinyl leukotrienes.
Early-onset allergic asthma is well controlled by corticosteroid therapy. In contrast, in late-onset eosinophilic asthma, severe symptoms are often recognized, and persistent sputum eosinophilia is observed despite treatment with inhaled and oral corticosteroids for several years. Thus, in this phenotype, a certain population of eosinophils may have acquired steroid resistance, and the eosinophils remaining in the lungs may be involved in the induction of the severe phenotype.1,2,4) On the other hand, non-Th2/type 2 asthma including obesity-related asthma and neutrophilic asthma are often steroid resistant and manifested by airway neutrophilia associated with increases in levels of the neutrophil chemoattractive cytokines interleukin (IL)-17 and IL-8 in the lung.1,2,4) More recently, it has been suggested that neutrophils are involved in the aggravation of asthma through “NETosis,” a specific cell death of neutrophils.5,6)
In this article, we review the pathogenesis of steroid-resistant asthma by focusing especially on neutrophils and NETosis. We hope that this review will be useful for the development of therapeutic drugs for the treatment of steroid-resistant asthma.
Asthma is regarded as a chronic airway disease based on Th2 inflammation. When a sensitized individual is exposed to a specific antigen, the antigenic protein penetrates into the airway mucosa through the epithelial barrier, followed by capture by antigen-presenting cells such as dendritic cells, which present the antigen peptide to memory Th2 cells (Fig. 1). Th2 cells are capable of producing the Th2 cytokines IL-4, IL-5, and IL-13, the main biological functions of which are the production of immunoglobulin E (IgE), recruitment of eosinophils into the local inflammatory site, and mucus production in the airway epithelium, respectively (Fig. 1). In addition, the chemotactic factor of eosinophils, IL-5, is produced from mast cells upon antigen-IgE antibody reaction on the cell membrane. Collectively, IL-5 is produced from both Th2 cells and mast cells in the lung and recruits eosinophils to the airways through the activation of IL-5 receptors on the cell membrane. The recruited eosinophils were demonstrated to be one of the major contributors to asthma pathogenesis, especially because neutralization of IL-5 by mepolizumab and blocking of the IL-5 receptor-α chain by benralizumab were demonstrated to be effective for the treatment of asthma in humans.7)
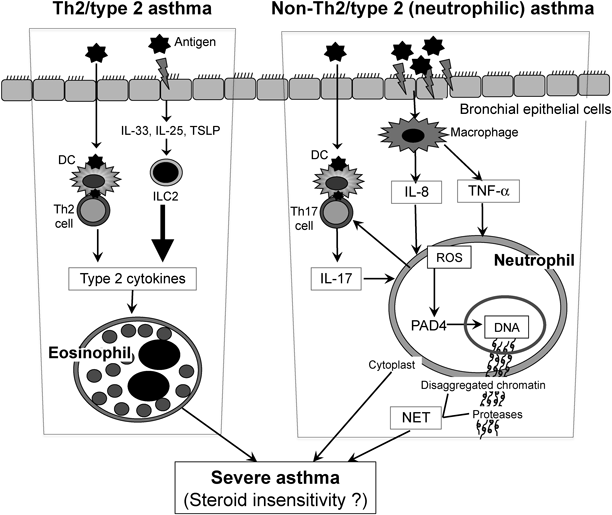
DC, dendritic cell; ILC2, type 2 innate lymphoid cell; NET, neutrophil extracellular trap; PAD4, peptidylarginine deiminase 4; ROS, reactive oxygen species; TSLP, thymic stromal lymphopoietin.
Generally, eosinophilic airway inflammation is well controlled by glucocorticoid therapy. However, in a certain patient population with severe asthma, classified as late-onset eosinophilic asthma, airway eosinophilia persists despite long-term corticosteroid therapy.1,2,4) It has recently been reported that many asthmatic patients have persistent airway type 2 inflammation even after glucocorticoid treatment, and that these patients are older and have more severe disease.8) However, the mechanisms of the steroid-resistant eosinophilic inflammation are unclear.
Recently, another important mechanism for allergic eosinophil infiltration into tissues has been reported.9–11) Type 2 innate lymphoid cells (ILC2) are capable of producing large amounts of IL-4, IL-5, and IL-13 in response to IL-33, IL-25, and thymic stromal lymphopoietin (TSLP), which are mainly produced from airway epithelial cells (Fig. 1). The levels of the type 2 cytokines from ILC2 were 10-fold greater than those from Th2 cells (Moro, unpublished data). Interestingly, it was reported that the IL-33-induced activation of ILC2 was resistant to glucocorticoid treatment in some allergic conditions.12) Thus, it is considered that the IL-33–ILC2–IL-5-eosinophil mechanism may be associated with the clinical pathogenesis of steroid-resistant eosinophilic asthma.
As reviewed by Wenzel1) and Kim et al.,4) non-Th2/type 2 asthma is characterized by neutrophilic airway inflammation. As summarized in Table 1, certain populations of both obesity-related asthma and neutrophilic asthma patients are resistant to glucocorticoid treatment. Although glucocorticoids suppress the apoptosis of neutrophils,13) there has been no direct evidence of how neutrophils are involved in the induction of steroid-resistant asthma. However, human and murine studies suggested that several cytokines including IL-17, IL-8, and tumor necrosis factor (TNF)-α are associated with the pathogenesis of steroid-resistant asthma.
Peripheral blood mononuclear cells isolated from some patients with steroid-resistant asthma produce high levels of IL-17A.2) In agreement with the clinical findings,2) Th17 cells adoptively transferred to immunodeficient mice challenged with antigen resulted in increased airway CXC chemokine secretion and neutrophilia, which were not inhibited by dexamethasone.14) In contrast, the transfer of Th2 cells resulted in airway eosinophilia that was significantly attenuated by dexamethasone.14) Therefore, IL-17 produced from Th17 cells recruit neutrophils into the lung, leading to steroid resistance in the model. As another subset of IL-17-producing cells, type 3 innate lymphoid cells (ILC3) are suggested to play roles in obesity-associated asthma,15) which is known to be steroid resistant.4)
IL-8 was also reported to be involved in steroid-resistant asthma. Multiple challenges with a pooled extract of dust mite, ragweed, and Aspergillus species in BALB/c mice resulted in neutrophilic airway inflammation, which was associated with nuclear factor-κB-mediated production of keratinocyte-derived chemokine (KC), a functional homologue of human IL-8.16) In agreement with that report,16) we also demonstrated that steroid-resistant asthma in a murine model coincided with neutrophilic airway inflammation, which was associated with increased levels of KC and macrophage inflammatory protein (MIP)-2, functional homologues of human IL-8 (Nabe, unpublished data). In the study, in ovalbumin (OVA) + Al(OH)3-sensitized BALB/c mice, intratracheal administration of a low dose of OVA produced marked eosinophilia and airway hyperresponsiveness, which were sensitively attenuated by dexamethasone. However, those responses induced by the administration of a 100-fold greater amount of OVA were resistant to the glucocorticoid. In addition, massive neutrophil infiltration into the lung as well as marked increases in levels of KC and MIP-2 were induced in the latter model developed with the higher OVA dose (Nabe, unpublished data). Taken together, the results indicate that IL-8 could also play important roles in the recruitment of neutrophils in steroid-resistant asthma.
In addition, increased TNF-α levels were observed in asthma patients who respond poorly to glucocorticoid therapy.17) In a murine study, TNF-α was reported to be involved in steroid-resistant asthma.18) When mice were sensitized with OVA + complete Freund’s adjuvant (CFA) and then challenged with OVA, steroid-resistant neutrophilic airway inflammation and an increase in TNF-α in the lung were induced, whereas the steroid-sensitive responses were normally induced in a standard OVA + Al(OH)3-sensitized model.18) Interestingly, neutralization of TNF-α canceled the steroid resistance in the OVA/CFA-sensitized model.18) Thus, it was concluded that TNF-α reduces the responsiveness to glucocorticoid in the neutrophilic airway inflammation model.18)
NETosis is a specific cell death of neutrophils.19–21) It is well known that neutrophils exert antibacterial function through the production of reactive oxygen species (ROS) in infectious conditions. In addition, neutrophils release their DNA outside cells, forming neutrophil extracellular traps (NETs), resulting in the exclusion of bacteria. Cell death for the formation of NETs is called “NETosis,” which is induced not only in infectious but also in noninfectious conditions.22) When NETosis is induced even in noninfectious conditions, the aggravation of inflammation could be induced through the mechanisms described below.22)
When neutrophils are exposed to infectious or noninfectious stimuli, ROS are produced and then activate peptidylarginine deiminase 4 (PAD4), which citrullinates histone in the nuclear chromatin chain resulting in disaggregation of the chain and release of chromatin in the extracellular space (Fig. 1). The extracellular chromatin is decorated with neutrophil proteases such as myeloperoxidase and elastase, forming NETs that induce inflammation22,23) (Fig. 1).
In a murine model of asthma, it has recently been reported that allergen-driven airway neutrophilia was decreased in PAD4-deficient mice with a decrease in NETosis.6) When sensitized mice were challenged with allergen and endotoxin, not only NETs but also enucleated neutrophil cytoplasts were increased in the lung. In the PAD4-deficient mice, NET formation and the increase in enucleated neutrophil cytoplasts together with the development of airway hyperresponsiveness were significantly decreased. However, degradation of NETs by instillation of deoxyribonuclease (DNase) did not produce neutrophilia. On the other hand, interestingly, the neutrophil cytoplasts activated lung dendritic cells in vitro to trigger antigen-specific IL-17 production from naïve CD4+ T cells. Therefore, it was suggested that neutrophil cytoplasts, but not NETs, were associated with Th17 cell-mediated neutrophilic airway inflammation in the murine model.6) In severe asthma patients, neutrophils together with NETs and cytoplasts were reported to be increased in the lung, and the increases were positively correlated with lung IL-17 levels.6) It will therefore be necessary to determine whether NETs and enucleated neutrophil cytoplasts are involved in the development of steroid-resistant asthma.
Glucocorticoids exert antiinflammatory action through the binding of glucocorticoid receptor (GR)α, which exists at the cytoplasm, binds hormone, translocates to the nucleus, and regulates gene transcription. On the other hand, GRβ, another isoform, which localizes at the nucleus, does not bind ligands and inhibits GRα function.24) It was reported that glucocorticoid insensitivity in asthma patients was associated with the loss of GRα nuclear translocation in bronchoalveolar lavage cells and elevated GRβ, which may inhibit GRα transactivation in response to steroids.25) Although GRs (both GRα and GRβ) were expressed in 100% of peripheral blood neutrophils, very low expression was observed in lung neutrophils in both healthy and chronic obstructive pulmonary disease (COPD) patients, suggesting that the lack of GR receptor expression in neutrophils in the airway tissues may contribute to glucocorticoid resistance in COPD patients.26)
Webster et al.27) demonstrated that treatment of HeLaS53 cells with TNF-α predominantly increased the expression of GRβ rather than GRα. Thus, the involvement of TNF-α in the development of steroid-resistant neutrophilic airway inflammation in mice described above18) may be associated with increased expression of GRβ in neutrophils. Although it had been accepted that GRβ did not exist in rodents,28) a more recent study has demonstrated that GRβ is expressed in mice in vivo and in vitro.29) Thus, further examination of the roles of GRβ in steroid-resistant asthma in murine studies is required.
Neutrophilic airway inflammation is one of the characteristic features of steroid-resistant asthma.1,2,4) Further elucidation of the mechanisms underlying the infiltration of neutrophils into the steroid-resistant asthmatic lung and the effector function is expected. Key molecules inducing neutrophilic airway inflammation may be targets of pharmacotherapy for the treatment of steroid-resistant asthma. For example, antibodies against IL-17, IL-8, and TNF-α are potential candidates.
On the other hand, because molecular-targeting drugs are generally expensive and may exert unknown side effects, drug repositioning studies should also be useful. For example, it was reported that macrolides such as azithromycin were effective in treating steroid-resistant asthma30,31) although the molecular mechanisms of the antiinflammatory effects are unknown. In addition, it was suggested that statins might restore glucocorticoid insensitivity in asthma.32) Preexisting drugs are relatively easy to reposition clinically because their pharmacological profiles have been extensively examined and their side effects can be anticipated.
The author declares no conflict of interest.