Abstract
Stroke is a common cerebrovascular disease. Inflammation-induced neuronal death is one of the key factors in stroke pathology. Propofol has been shown to ameliorate neuroinflammatory injury, but the exact mechanism of its neuroprotective role remains to be fully elucidated. In the present study, we found that inflammation was activated in ischemic cortical neurons, and the expression of nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing 1 (NLRP1), NLRP3 inflammasome and effectors in primary cortical neurons increased. However, we found that propofol could inhibit the increased expression of NLRP1 and NLRP3 inflammasome induced by oxygen-glucose deprivation (OGD). Furthermore, the effector molecule caspase-1 (casp1) was revealed to be the downstream target of NLRP1 and propofol repressed the activation of caspase-1 via inhibiting NLRP1 in cortical neurons. Moreover, propofol inhibits caspase-6 activation in neurons through the NLRP1-caspase-1 pathway. Once the expression of caspase6 increases, propofol reduced its neuroprotective effect in OGD-treated cortical neurons. In the stroke middle cerebral artery occlusion (MCAO) model, infusion of caspase-6 inhibitors enhanced the protective effect of propofol on infarct size and neurological function. In conclusion, our results suggest that propofol plays a neuroprotective role in stroke by inhibiting the inflammatory pathway of NLRP1-caspase-1-caspase-6. Overall, these data suggest that propofol plays a key role in the inflammatory-dependent pathway after stroke, providing an important evidence for propofol as an effective strategy for neuroprotection in stroke.
INTRODUCTION
Stroke is a disease caused by cerebral vascular obstruction and/or rupture which induces brain damage and leads to permanent disability and death.1,2) The ischemic stroke is 80–85% of total stroke.3) Various risk factors are involved in the pathogenesis for the neurons of ischemic stroke, including inflammation, oxidative stress and excitotoxicity.4) Accumulating evidence suggests that the immune system plays an important role in the development of nervous system diseases.5) Studies have found that the immune system-related inflammatory processes are closely associated with stroke.6) Activation of immune response induces the expression of pro-inflammatory cytokines, chemokines and reactive oxygen species, which triggered the inflammatory response of the nervous system.7) As an important part of the innate immune system, inflammasome plays a pivotal role in inflammatory cascade.8) This incites us to study neuronal responses to inflammation and to link inflammation to neuronal molecular events associated with stroke.
Inflammasome are macromolecular complexes, which are usually composed of the nucleotide-binding oligomerization domain-like receptor (NLR) molecules, the adaptor protein apoptosis speck-like protein with a caspase recruitment domain (ASC) and the effector molecule caspase-1 (casp1).9) It has been reported nucleotide-binding domain, leucine-rich-repeat containing family, pyrin domain-containing (NLRP) inflammasomes were tightly involved in the pathogenesis of stroke. NLRP1 was revealed to increase due to the decreased miR-9a-5p induced by stroke, resulting in the activation of inflammatory response and subsequent neuron death.10) NLRP2 was also high-expressed in astrocytes of an ischemic stroke mouse model.11) Furthermore, NLRP3 was positively modulated via nuclear factor-kappaB (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways in ischemic stroke-treated neurons.12) Currently, many evidences demonstrated that inflammasome can regulate the activation of casp1 via proximity-induced auto-activation.13) Casp1 is a cysteine peptidase involved in apoptosis, which causes inflammation and cell apoptosis by hydrolyzing pro-inflammatory cytokine protein in mature interleukin-1 beta (IL-1β).14) In Alzheimer’s disease, increased casp1 was reported to induce caspase-6 (casp6) activation. Meanwhile, casp6 activity is associated intimately with formalin-induced pain pathology and inflammation inhibition.15) In ischemic stroke models, casp6 was activated in human primary neurons deprived of serum16) and casp6 selectively participates in axonal mutagenesis and neuronal apoptosis.17) Though inflammation response participates in the development of stroke, it remains to be illustrated whether casp1 can regulate casp6 activation in this process.
Propofol is an intravenous drug widely used in surgery and severe sedation.18) In recent years, the neuroprotective effect of propofol has been found. The potential mechanisms of propofol’s neuroprotective role include inhibition of pro-inflammatory factors, antioxidation, regulation of apoptotic signaling and activation of neuroprotective pathways.19,20) In traumatic brain injury model, propofol was reported to ameliorate cortical injury by inhibiting the expression and activation of NLRP3 inflammasome in the cerebral cortex.21) In a rat model of pain, propofol relieved the inflammation response via negatively modulating NLRP3 expression. In stroke, propofol tended to decrease the aggregation of α-synuclein and alleviate the neuronal injury. Also, propofol repressed the excessive activation of microglia through the A2b receptor and protected neurons from the inflammatory injury following cerebral infarction. It’s widely acknowledged propofol exerts a anti-inflammation role in central nerve system. The inhibitory effect of propofol in neuroinflammation makes it an attractive candidate for the treatment of stroke. However, little is known whether propofol regulate the expression of NLRP1 or NLRP3 inflammasome in stroke. The underlying neuroprotective mechanism of propofol in stroke remains to be further explored.
In this study, we investigated the role and potential molecular mechanisms of propofol in cell and animal stroke models. We found that NLRP1 and NLRP3 were increased in cortical neurons after oxygen-glucose deprivation (OGD) exposure while propofol treatment compromised the effect of OGD exposure. Furthermore, casp1 was downstream target of NLRP1 and propofol could repress OGD-induced casp1 activation by inhibiting the expression of NLRP1. Moreover, casp6 was regulated by casp1 in OGD-treated neurons and propofol can suppress casp6 activation by inhibiting NLRP1-casp1 pathway. In stroke mice, propofol was found to protect cortical neurons by inhibiting casp6 activity, reduce infarct size and improve neurological function. In general, our results suggest that propofol can ameliorate neurological injury after stroke by inhibiting the activation of NLRP1-casp1-casp6 inflammatory pathway. Our study provides a promising strategy for the treatment of ischemic stroke.
MATERIALS AND METHODS
AnimalsIn the experiment, male C57BL/6J mice aged 5–7 weeks were treated according to the laboratory animal care guidelines. All experimental animal protocols were approved by the committees of China-Japan Union Hospital of Jilin University. The mice were kept in a 12-h light–dark cycle, allowing free access to food and water.
Primary Cortical Neuronal CulturesCultured primary cortical neurons were prepared from 16-d C57B/6 mouse embryos.22) Briefly, Mouse fetuses were collected in cold HBSS medium (Fisher Scientific, Hanover Park, IL, U.S.A.). The fetal brains were removed and their cortex tissues were collected in cold HBSS. Cells and tissues were dissociated with 2.5% trypsin in HBSS while shaking in a 37 °C water bath. Cell were cultured in 35 mm culture dishes containing penicillin and streptomycin (50 U/mL medium), glutamine (2 mM), glucose (25 mM), 2% B27 serum-free supplement and maintained at 37°C in 95% air and 5% CO2 incubator.
Oxygen Glucose DeprivationIn order to simulate ischemic state, neurons were treated with oxygen-glucose deprivation. Neurons were washed twice in a glucose-free equilibrium salt solution and incubated in a hypoxic chamber at 37°C for 3, 6, and 12 h, respectively. Control group was cultured under normal oxygen in a balanced salt solution containing glucose. After OGD culture, the glucose-free medium was replaced by fresh neuron culture medium and placed in the conventional culture room for 24 h. Then, propofol (40 μM) or Z-YVAD-fmk (casp1 inhibitor, 5 μM, selleck) was added respectively to the cells 1 h before and during the hypoxia-reoxygenation.
Western Blot AnalysisThe expression levels of NLRP1, ASC, casp1 and casp6 were detected by Western blotting. Ultrasound was used to prepare the whole-hole extract of cortical neurons, and centrifugal concentration was carried out under the condition of 12000 × g to obtain the supernatant. Bicinchoninic acid (BCA) protein detection kit was used to determine the concentration of total protein. Each protein sample (30 μg) was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gels and transferred to polyvinylidene fluoride (PVDF) (Thermo Fisher Scientific, U.S.A.). At room temperature, 5% (w/v) dry skim milk was used and sealed in Tris buffer for 1 h. The membrane was incubated overnight with NLRP1 (1 : 500, Abcam, U.S.A.), ASC (1 : 500, Bioworld, U.S.A.), caspase-1 (1 : 500, Bioworld), caspase 6 (1 : 500, Bioworld) and actin (1 : 1000) at 4°C. The second antibody(1 : 2000, Wuhan Boster Biological Technology, Ltd., Wuhan, China) combined with HPR was incubated at room temperature for 1 h and washed three times with buffer. The film was reacted with the enhanced luminescence solution and the film was imaged. The protein was quantitatively analyzed by image J software, and the normalized relative density of target protein was calculated.
Cell TransfectionNLRP1, casp1, casp6 overexpression and vector plasmid were purchased from Genechem (Shanghai, China). The cells were seeded in 6-well plates and fused in 70–80%. According to the manufacturer’s instructions, the construct was transported into 1 × 106 cortical neurons at a concentration of 4 μg by Lipofectamine 2000. Cells were collected and their protein expression was analyzed 24 h after transfection.
Caspase Activity AssaysAt the specified time, the cells were collected and suspended in Buffer lysing, incubated on ice for 10 min, and centrifuged for 3 min under 250 × g conditions to collect the cytoplasmic extract. The extract was mixed with the mixture of casp reaction, which contained 20 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 0.1% CHAPS, pH 7.4, 10 mM dithiothreitol (DTT), 2 mM ethylenediaminetetraacetic acid (EDTA) and 5% sucrose, and either 10 mM AC YVAD AFC reacted with casp1 or AC VEID AFC reacted with casp6. The fluorescent microplate reader was used to measure the activity at a wavelength of 380 and 505 nm.
Enzyme-Linked Immunosorbent Assay (ELISA)As described in Western blot section, the cortical neurons were resuspended with lysis buffer. And we collected the supernatant after centrifugation. Part of the supernatant was analyzed using BCA protein detection kit to determine the protein concentration. Other supernatant was retained for the ELISA cell experiments. ELISA was performed according to the protocols of manufacturer. Human IL-1β ELISA kit (EK0392) and tumor necrosis factor (TNF)α ELISA kit (EK0525) were purchased from Boster biological company.
Assessment of Cell DeathCells were placed in 96 plate orifice plate and cultured in 37°C incubator. Cell survival analysis was carried out with Cell Counting Kit-8 (CCK-8; Sigma-Aldrich). In the measurements, 10 μL CCK-8 was added to each well, according to the manufacturer’s instructions, and optical density (OD) value was measured at 450 nm using a microplate reader. Quantitative assessment of cell injury by measuring lactate dehydrogenase (LDH) in culture medium. According to the manufacturer’s protocol, LDH Cytotoxicity Detection Kit was used to detect the amount of LDH released into culture medium 24 h after OGD. Read the results on a microplate reader.
Animal ModelThe experimental stroke model mice were induced by middle cerebral artery occlusion (MCAO) and reperfused to C57BL/6J mice. All mice were evaluated with the standard of neurological deficits before constructing the MCAO model. Mice with neurologic deficit (including 1, 2, 3, and 4 point assessed in the neurologic deficit evaluation) were excluded from the experiments. During the operation, the animal’s body temperature is maintained at about 37 °C by a heating pad. The mice were anesthetized with 0.4% sodium pentobarbital (40 mg/kg, intraperitoneally (i.p.)) and fixed in supine position. The common carotid artery, external carotid artery and internal carotid artery were separated after carotid artery was exposed through a midline incision. The 6.0 nylon monofilament coated with standardized silicone rubber was pushed into the right internal carotid artery 9–10 mm from the stump of the external carotid artery to block the origin of the middle cerebral artery. After 1 h of ischemia induction, the occlusive filament was withdrawn and the ischemic brain area was reperfused for 24 h. The mice were infused with saline, propofol (10 mg/mL, intravenously (i.v.)) or Z-VEID-fmk (caspase-6 inhibitor, 50 μM, Apexbio, U.S.A.) in the initial 30 min of reperfusion. After 24 h of perfusion, mice in each experimental group were perfused with polyformaldehyde (4%) after cold saline was perfused into the heart. Brain tissue was immediately removed and cut into 2 mm coronal sections. The brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride for 15 min and fixed on 4% polyformaldehyde. Normal tissues were stained red and infarcted tissues were not stained (white). Digital image J (Ver1.37c, NIH) was used to calculate the percentage of infarct area in each slice, and the infarct volume was calculated by multiplying the thickness of the interval wall by the percentage of the whole hemisphere.
Evaluation of Neurological DeficitAfter 24 h of perfusion, based on the Longa et al.23) standard, neurological deficits were assessed by 5-point scale system in a blinded fashion. The following are carried out: 0, no neurologic deficit; 1, the right forelimb is not fully extended; 2, circling to right; 3, no autonomic movement on the left side; 4, no spontaneous walking; the more serious the motor injury, the higher the score of nerve function defect.
Statistical AnalysisStatistical analysis of data was performed using GraphPad Prism software (La Jolla, CA, U.S.A.). Except for the neurological deficit score, the data were expressed as the standard error of the average value. Student t-test or ANOVA and Tukey post hoc test were used to test and analyze the data. Kruskal–Wallis test and Mann–Whitney U test were used for multiple comparative analysis of neurological deficit score. p < 0.05 was considered to have statistical significance.
RESULTS
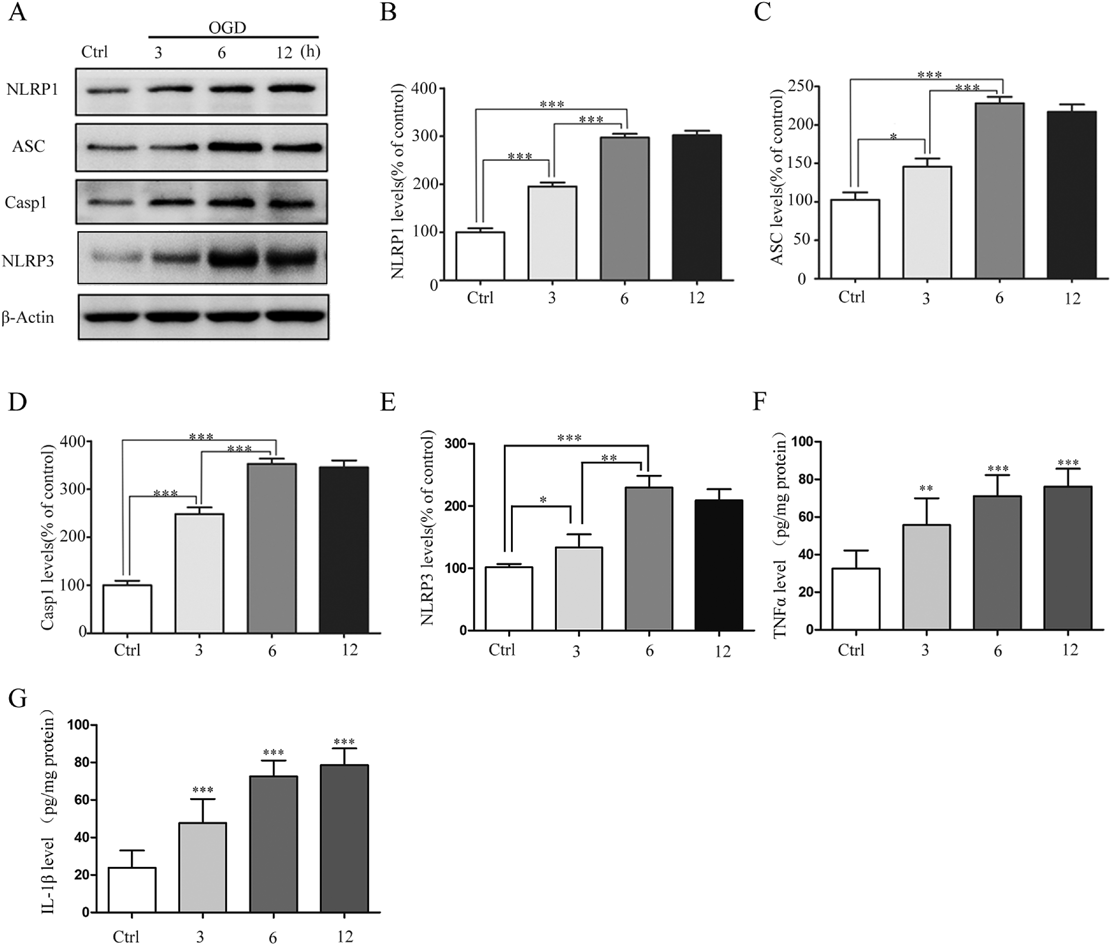
Increased Expression of NLRP1 and NLRP3 Inflammasome in Ischemic Primary Cortical NeuronsTo determine whether simulated ischemic condition activated the NLRP1 and NLRP3 inflammasome in primary cortical neuron, we analyzed the expression of NLRP1 and NLRP3 inflammasome elements in primary cortical neuron exposed to OGD. The levels of NLRP1, NLRP3, ASC and casp1 protein were detected in primary cortical neuron by Western blotting (Figs. 1A–E). The result showed that the levels of NLRP1 and NLRP3 inflammatory components and effectors in primary cortical neurons increased after 3 h of treatment with OGD, then reached the highest levels at 6 h, and maintained unchanged in 6–12h. To investigate the effect of OGD exposure on inflammation response, we measured the production of TNFα and IL-1β of cortical neurons after OGD treatment. The result showed that TNFα and IL-1β increased after OGD treatment in a time-dependent manner (Figs. 1F, G). The results showed that NLRP1 and NLRP3 inflammasome in primary cortical neurons were activated after exposure to OGD.
Propofol Repressed the Activation of Casp1 via Inhibiting NLRP1We next evaluated the potential anti-inflammatory property of propofol in primary cortical neuron subjected to simulated ischemia. Firstly, we explored the effect of different dose of propofol on OGD-treated cortical neurons. The result indicated that propofol treatment decreased the expression of NLRP1 and NLRP3 in a dose-dependent manner and 40 μM was able to induce an obvious change. So 40 μM propofol was determined to treat neurons in later experiments (Figs. 2A–C). Next, we treated neurons with 40 μM propofol before OGD exposure. Similarly, the result showed that compared with control group, NLRP1 and NLRP3 expression increased after exposure to OGD, while the level of NLRP1 and NLRP3 decreased in neurons treated with propofol (Figs. 2D–F). NLRP1 has been reported to regulate the expression of casp1 in cortical neurons and we wanted to know whether propofol treatment affected the expression of casp1 via regulating NLRP1 in OGD-treated neurons. To explore the linkage between propofol and casp1, we overexpressed and verified the expression of NLRP1 in neurons firstly (Fig. 2G). Then we measured propofol-induced caspase expression and activities after OGD exposure and NLRP1 overexpression. The results suggested that casp1 expression and activity in primary cortical neurons increased after the treatment of OGD, but treatment with propofol induced inhibition of casp1 expression and activity compared with OGD group. Moreover, overexpression of NLRP1 reversed the inhibitory effect of propofol to a certain extent and restored the expression and activity of casp1 (Figs. 2H–J). In conclusion, these results indicate that casp1 was activated after OGD treatment and the propofol restrained casp1 activation in cortical neuron partly by inhibiting NLRP1.
Propofol Suppressed Casp6 Activation by Inhibiting NLRP1–Casp1 PathwayIt has been found that casp6 is involved in regulating neuronal apoptosis and axonal degeneration apoptosis of neurons.16) To verify whether casp6 is involved in the stroke pathogenesis, we measured the expression and activity of casp6 after exposure to OGD. To explore the detailed molecular mechanism, we overexpressed NLRP1 and casp1 respectively when treated with propofol and OGD. The result hinted that casp1 was highly expressed when transfected with casp1 plasmid (Fig. 3A). Compared with the control group, casp6 expression and activity were enhanced in cortical neurons treated with OGD. We next investigated whether propofol influenced the expression and activity of casp6, the results showed that propofol treatment reversed the effect of OGD on the expression and activity of casp6 in cortical neurons. Furthermore, we found that over-expression of NLRP1 and casp1 compromised the inhibitory effect of propofol on casp6 expression and activity (Figs. 3B–D). These data confirmed that casp6 was activated in OGD-treated neurons and propofol inhibited casp6 activation partly through NLRP1-casp1 pathway.
NLRP1 Was Essential to Mediate the Inhibitory Effect of Propofol on the Inflammatory Activities of Cortical NeuronsTo determine the critical role of NLRP1 in mediating the inflammatory-inhibitory role of propofol in OGD-treated neurons, we constructed the NLRP1 knockdown neurons using lentiviral vector carrying NLRP1 interference sequences. The NLRP1 expression was obviously decreased when infected with the lentiviral vector (Figs. 4A, B). Then we treated NLRP1 NC and KD neurons with propofol before OGD exposure. The result showed that casp1 and casp6 were decreased in NLRP1 KD neurons when subjected to OGD insult compared with NC neurons exposed to OGD (Figs. 4C–F). To determine the critical role of NLRP1 in inflammation factors production, we measured the expression of tumor necrosis factor-alpha (TNFα) and IL-1β. The result showed that TNFα and IL-1β increased when subjected to OGD insult. However the pretreatment of propofol reversed the increase of TNFα and IL-1β. Compared with NC neurons, TNFα and IL-1β were obviously reduced in NLRP1 KD neurons when exposed to OGD treatment (Figs. 4G, H). These results indicated that NLRP1 is critical to induce the inflammation response and mediated the anti-inflammatory effect of propofol in stroke.
Propofol Protects Cortical Neurons and Brain Tissues by Inhibiting Casp6 Activity under in Vitro and in Vivo Ischemic ConditionsWe tested in vitro whether propofol plays a neuroprotective role in stroke. CCK8 and LDH were used to measure the neuronal activity. Compared with the control group, the neuronal death induced by OGD increased significantly, while propofol treatment reduced OGD-induced cell death. For casp6 was the downstream target of NLRP1 and casp1, we manipulated the expression or activity of casp6 in following experiments. The result showed that overexpression of casp6 reversed the protective effect of propofol (Figs. 5A, B). Next, we evaluated the potential effect of propofol in ischemic stroke mice. Compared with the sham-operated group, the neurological function of ischemic perfusion mice was severely damaged. The neurological function of MCAO mice treated with propofol or Z-VEID-fmk (casp6 inhibitor) was improved. Then, compared with propofol or casp6 group separately, the neurological score of MCAO mice treated with propofol and casp6 inhibitor decreased significantly, indicating a better neurological function (Fig. 5C). Coronal section images showed that severe infarction occurred in the right brain of mice treated with MCAO compared with mice treated with sham operation. Similarly, propofol intravenous infusion significantly reduced infarct volume in stroke mice, and the same inhibitory effect was found in casp6 inhibitor infusion mice. Furthermore, compared with propofol-treated MCAO mice, casp6 inhibitor infusion after stroke can enhance the protective effect of propofol on stroke-induced injury, showing a smaller infarct volume (Figs. 5D, E). Our results confirm that casp6 is one of the downstream target of propofol and propofol protects cortical neurons and brain tissues during ischemia in vitro and in vivo.
DISCUSSION
Stroke is one of the major causes of death and disability worldwide.24) Many mechanisms are individually or in combination involved in the progression of stroke, including excitotoxicity, neuroinflammation, oxidative stress and programmed cell death.25) In particular, inflammatory cascade reaction is considered to be a key factor in the progression of ischemic stroke.26) Recently, the inhibitory effect of propofol in neuroinflammation makes it an attractive candidate for the treatment of stroke. However, the potential mechanism of propofol in neuroprotection is not fully understood. In the present study, we found that NLRP1 inflammasome were activated in cerebral ischemic cortical neurons. Propofol treatment not only inhibited the increased expression of NLRP1 after exposure to OGD, but also repressed the activation of casp1 by inhibiting the expression of NLRP1. In addition, propofol can also inhibit the activity of casp6 by inhibiting NLRP1-casp1 pathway, which affects the survival of cortical neurons. In stroke animal models, infusion of casp6 inhibitors enhanced the protective effect of propofol on infarct size and neurological function. Our work revealed NLRP1-casp1-casp6 pathway is involved in the pathogenesis of stroke, and propofol plays a neuroprotective role by inhibiting this inflammatory pathway.
One marker of inflammatory response is the activation of inflammasomes. Then, activation of inflammasome complexes contributes to inflammation in neurologic diseases and leads to neuronal death.27) In this study, we established a well characterized OGD stroke model and tested it. It was previously reported that the expression of NLRP1 and NLRP3 increased in a time-dependent manner in oxygen/glucose-inhibited cortical neurons.28) Moreover, in vivo and in vitro models of ischemic stroke, the expression of NLRP2 protein is increased.11) Our results showed that the levels of NLRP1 and NLRP3 protein and casp1 increased in cortical neurons after OGD exposure, which was consistent with previously published work. OGD exposure leads to apoptosis of human cortical neurons, which may be related to altered expression of inflammasomes.29) Indeed, our work revealed that exposure of cortical neurons to OGD markedly increased inflammatory cytokines, which may lead to neuronal cell death and exacerbate ischemic brain injury. Our data indicate that NLRP1 inflammasomes may be involved in stroke-induced neuronal death.
The increased production of pro-inflammatory cytokines contributes to and exacerbates ischemic brain injury. Propofol, a commonly used intravenous anesthetic,18) has therapeutic effects on neuroinflammation in various brain injury models. Recent studies have suggested that propofol ameliorates blast-induced traumatic brain injury by inhibiting the expression and activation of NLRP3 inflammasome and pro-inflammatory cytokines maturation.21) In the animal model of ischemia-reperfusion injury, propofol provides neuronal protection by inhibiting the expression of basic fibroblast growth factor (bFGF).30) However, the effect of propofol on the inflammasome is unknown and remained to be explored. Therefore, the main focus of this study was to test whether propofol ameliorated neuronal injury of stroke, at least to some extent, by acting on inflammatory-dependent pathway. Indeed, in this study, we found that propofol treatment significantly decreased the expression NLRP1 and NLRP3 in OGD-treated neurons. These results revealed that propofol may exert its neuroprotective effect by inhibiting the inflammasome of NLRP1 and NLRP3.
Inflammation cascade is involved in the complex mechanism of brain injury after ischemia. Indeed, previous studies have shown that inhibition of NLRP1 inflammation leads to the decrease of casp1 activity.9) Activation of casp1 leads to the activation and maturation of pro-inflammatory cytokine IL-1β, which plays a key role in the inflammatory pathway.31) Our work indicated that treatment with propofol inhibited the activation of casp1, and increased expression of NLRP1 blocked the inhibitory effect of propofol on casp1 activation. To further validate that propofol exert its anti-inflammatory role via targeting NLRP1. We tested the effect of propofol in NLRP1 knockdown neurons. The result showed that OGD treatment failed to increase casp1 and propofol lose its anti-inflammatory role in NLRP1 knockdown neurons. These in vitro findings suggest that propofol inhibits the activation of casp1 in cortical neurons by inhibiting NLRP1.
Previous studies showed that casp6 is elevated and activated after excitotoxic injuries of primary rat neurons.32) inhibition of casp-1 can block downstream activation of casp6.33) However, it remains to be explored whether casp1 regulates casp6 activity in stroke models. In the present study, we found the expression of casp6 increased in response to OGD. Treatment with propofol could significantly reduce casp6 activity while overexpression of NLRP1 or casp1 could reversed casp6 activity, indicating casp6 mediated the therapeutic effect of propofol in stroke. The previous studies have shown that active casp-6 is closely related to apoptosis of human neurons.34) Casp6 gene deletion resulted in improved neurological function and reduced neuronal death.35) Thus we speculated that the neuroprotective effect of propofol was associated with reduced expression of casp6. Our experiment showed that propofol treatment can reduce OGD-induced cell death and plays a neuroprotective role in stroke. While casp6 overexpression reversed the protective effect of propofol and led to increased neuronal death. The animal experiment results revealed that infusion of casp6 inhibitors could significantly enhance the protective effect of propofol on stroke-induced injury, showing smaller infarct size and lower neurological score. Based on these data, we propose that casp6 as a critical regulator in neuronal apoptosis cascade induced by cerebral ischemia.
However, there are still some issues remained to be explored in the future study. Dose propofol target NLRP1 directly or indirectly? Previous studies have reported that propofol regulated NLRP3 inflammasome via inhibiting reactive oxygen species (ROS) or nuclear factor kappaB (NF-κB) in acute brain or liver injury.21,36) It is possibly that propofol inhibits NLRP1 via targeting ROS or NF-κB in stroke. For the lack of agonist of casp6, we used casp6 inhibitor to verify the role of casp6 in mediating the effect of propofol in stroke. Actually, it’s better to construct the casp6 overexpressed transgenic mouse to examine this conclusion.
In our study, we found that exposure to OGD could induce the expression of NLRP1 and NLRP3 in cortical neurons while propofol reduced the up-regulation of NLRP1 and NLRP3 induced by OGD. Furthermore, propofol could repress the activation of casp1 by inhibiting the expression of NLRP1. In addition, propofol suppressed casp6 activation by inhibiting NLRP1–casp1 pathway. In vitro, we confirmed that propofol promoted the survival of neurons through inhibiting NLRP1-casp1–casp6 pathway. In vivo, we confirmed that infusion of casp6 inhibitors can enhance the protective effect of propofol on stroke-induced injury, resulting in smaller infarct size and significant improvement of neurological deficits. Our study provided novel potential strategy for the treatment of stroke.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav., 87, 179–197 (2007).
- 2) Hou ST, MacManus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int. Rev. Cytol., 221, 93–148 (2002).
- 3) Zhang GL, Zhu ZH, Wang YZ. Neural stem cell transplantation therapy for brain ischemic stroke: review and perspectives. World J. Stem Cells., 11, 817–830 (2019).
- 4) Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke, 40, e331–e339 (2009).
- 5) Kenney MJ, Ganta CK. Autonomic nervous system and immune system interactions. Compr. Physiol., 4, 1177–1200 (2014).
- 6) Jiang C, Kong W, Wang Y, Ziai W, Yang Q, Zuo F, Li F, Wang Y, Xu H, Li Q, Yang J, Lu H, Zhang J, Wang J. Changes in the cellular immune system and circulating inflammatory markers of stroke patients. Oncotarget, 8, 3553–3567 (2017).
- 7) Hernangómez M, Carrillo-Salinas FJ, Mecha M, Correa F, Mestre L, Loría F, Feliú A, Docagne F, Guaza C. Brain innate immunity in the regulation of neuroinflammation: therapeutic strategies by modulating CD200-CD200R interaction involve the cannabinoid system. Curr. Pharm. Des., 20, 4707–4722 (2014).
- 8) Amantea D, Micieli G, Tassorelli C, Cuartero MI, Ballesteros I, Certo M, Moro MA, Lizasoain I, Bagetta G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci., 9, 147 (2015).
- 9) de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow Metab., 29, 1251–1261 (2009).
- 10) Yuze C, Huixue Z, Xiaoyu L, Jianjian W, Xiaoming Z, Shengnan S, Zhonglei B, Wenqi T, Shangwei N, Lihua W, Liying C. Overexpression of MicroRNA-9a-5p ameliorates NLRP1 inflammasome-mediated ischemic injury in rats following ischemic stroke. Neuroscience, (2020), in press.
- 11) Sun X, Song X, Zhang L, Sun J, Wei X, Meng L, An J. NLRP2 is highly expressed in a mouse model of ischemic stroke. Biochem. Biophys. Res. Commun., 479, 656–662 (2016).
- 12) Fann DY, Lim YA, Cheng YL, Lok KZ, Chunduri P, Baik SH, Drummond GR, Dheen ST, Sobey CG, Jo DG, Chen CL, Arumugam TV. Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol., 55, 1082–1096 (2018).
- 13) Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol., 28, 137–161 (2012).
- 14) Mastronardi C, Whelan F, Yildiz OA, Hannestad J, Elashoff D, McCann SM, Licinio J, Wong ML. Caspase 1 deficiency reduces inflammation-induced brain transcription. Proc. Natl. Acad. Sci. U.S.A., 104, 7205–7210 (2007).
- 15) Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lü N, Liu YC, Ji RR. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J. Clin. Invest., 124, 1173–1186 (2014).
- 16) Shabanzadeh AP, D’Onofrio PM, Monnier PP, Koeberle PD. Targeting caspase-6 and caspase-8 to promote neuronal survival following ischemic stroke. Cell Death Dis., 6, e1967 (2015).
- 17) Zhang Y, Goodyer C, LeBlanc A. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and -8. J. Neurosci., 20, 8384–8389 (2000).
- 18) Chidambaran V, Costandi A, D’Mello A. Propofol: a review of its role in pediatric anesthesia and sedation. CNS Drugs, 29, 543–563 (2015).
- 19) Marik PE. Propofol: an immunomodulating agent. Pharmacotherapy, 25, 28S–33S (2005).
- 20) Yu Y, Jian MY, Wang YZ, Han RQ. Propofol ameliorates calpain-induced collapsin response mediator protein-2 proteolysis in traumatic brain injury in rats. Chin. Med. J. (Engl.), 128, 919–927 (2015).
- 21) Ma J, Xiao W, Wang J, Wu J, Ren J, Hou J, Gu J, Fan K, Yu B. Propofol inhibits NLRP3 inflammasome and attenuates blast-induced traumatic brain injury in rats. Inflammation, 39, 2094–2103 (2016).
- 22) Madineni A, Alhadidi Q, Shah ZA. Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol. Neurobiol., 53, 867–878 (2016).
- 23) Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke, 20, 84–91 (1989).
- 24) Bousser MG. Platelets, atherothrombosis, antiplatelet drugs and cerebral ischemia. Bull. Acad Natl. Med., 197, 389–394 (2013), Platelets, atherothrombosis, antiplatelet drugs and cerebral ischemia.
- 25) Turley KR, Toledo-Pereyra LH, Kothari RU. Molecular mechanisms in the pathogenesis and treatment of acute ischemic stroke. J. Invest. Surg., 18, 207–218 (2005).
- 26) Esenwa CC, Elkind MS. Inflammatory risk factors, biomarkers and associated therapy in ischaemic stroke. Nat Rev Neurol., 12, 594–604 (2016).
- 27) Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem., 136 (Suppl. 1), 29–38 (2016).
- 28) Fann DY, Lee SY, Manzanero S, et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis., 4, e790 (2013).
- 29) Gong Z, Pan J, Shen Q, Li M, Peng Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflammation, 15, 242 (2018).
- 30) Zhao XC, Zhang LM, Tong DY, An P, Jiang C, Zhao P, Chen WM, Wang J. Propofol increases expression of basic fibroblast growth factor after transient cerebral ischemia in rats. Neurochem. Res., 38, 530–537 (2013).
- 31) Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell, 117, 561–574 (2004).
- 32) Girling KD, Demers MJ, Laine J, Zhang S, Wang YT, Graham RK. Activation of caspase-6 and cleavage of caspase-6 substrates is an early event in NMDA receptor-mediated excitotoxicity. J. Neurosci. Res., 96, 391–406 (2018).
- 33) Martin DDO, Schmidt ME, Nguyen YT, Lazic N, Hayden MR. Identification of a novel caspase cleavage site in huntingtin that regulates mutant huntingtin clearance. FASEB J., 33, 3190–3197 (2019).
- 34) Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol., 165, 523–531 (2004).
- 35) Harrison DC, Davis RP, Bond BC, Campbell CA, James MF, Parsons AA, Philpott KL. Caspase mRNA expression in a rat model of focal cerebral ischemia. Brain Res. Mol. Brain Res., 89, 133–146 (2001).
- 36) Zhang Z, Tian L, Jiang K. Propofol attenuates inflammatory response and apoptosis to protect d-galactosamine/lipopolysaccharide induced acute liver injury via regulating TLR4/NF-kappaB/NLRP3 pathway. Int. Immunopharmacol., 77, 105974 (2019).