Current Topics: Reviews
Selenoprotein P; P for Plasma, Prognosis, Prophylaxis, and More
2020 Volume 43 Issue 3 Pages 366-374
Details
2020 Volume 43 Issue 3 Pages 366-374
Selenoprotein P (SeP) is one of the 25 human selenocysteine (Sec)-containing proteins, and is generally thought to function as a plasma carrier of the trace element selenium in the body. Recent studies, however, indicate unsuspected pivotal roles of SeP in human diseases, particularly in type 2 diabetes mellitus (T2DM) and pulmonary arterial hypertension (PAH). In this review, we will summarize the characteristics of SeP and recent advances in the field, especially focusing on the emerging roles of SeP in pathophysiological conditions. We will also discuss potential medical/pharmaceutical applications targeting SeP.
Selenoproteins are defined by the presence in the polypeptides of at least one selenocysteine (Sec) residue, the 21st codon-encoded amino acid, and can be found in many kinds of organisms, from bacteria to mammals.1) The human genome encodes at least 25 selenoproteins, including well-characterized glutathione peroxidases (GPxs) and thioredoxin reductases (TrxRs). Consistent with the high nucleophilic reactivity of a selenol group (-SeH) in Sec, compared to a thiol group (-SH) in cysteine (Cys), many selenoproteins exhibit redox-related enzymatic activities, with Sec residues at their catalytic centers. Indeed, some of these selenoenzymes play essential roles in cellular reactive oxygen species (ROS)-scavenging systems.1) Thus, a well acknowledged primary function of the selenoprotein family is the efficient protection of the body from such stresses, suggesting indispensable roles in maintaining cellular/tissue homeostasis.
Selenoprotein P (SeP) was first described in 1973,2,3) and purification of the protein was reported in 1982 as the major selenoprotein in rat plasma, to which more than half of plasma selenium (Se) was attributed.4,5) SeP is a unique selenoprotein, possessing a large number of Sec residues (10 Sec codons in-frame in human cDNA),6,7) whereas other members typically bear one.1) This feature confers SeP a function as the predominant and highly efficient carrier that delivers Sec to the whole body, especially to the brain and testis, serving as a cellular Se resource for the de novo synthesis of other essential selenoproteins. Together with its phospholipid peroxidase activity, SeP is therefore thought to play central roles in selenoprotein-mediated redox homeostasis. Thus, low plasma SeP would disturb such redox balances and, in fact, is reported to be related to several diseases. On the other hand, recent studies have revealed that increased plasma SeP is also associated with other human disorders. More importantly, several studies, including ours, surprisingly specified that elevated SeP is not merely a consequence of, but has profound causal and/or promoting effects on, the progression of some diseases. These studies present an emerging potential of SeP as a therapeutic/pharmaceutical target, in addition to being a biomarker for prediction or diagnoses. In this review, we will first briefly summarize characteristics of SeP, since excellent reviews have been published,8–10) and will then provide an overview of recent advances in SeP research, especially focusing on relationships between SeP and some human diseases, as well as the potential medical/pharmaceutical application of SeP.
The human SeP-coding gene allele, SELENOP, is mapped as 5p11 in the latest version of the human genome (GRCh38.p13); the mRNA has a total of 10 UGA “opal” codons in-frame which usually function as one of the stop codons, followed by its bona fide termination codon UAA and 3′UTR. Two peculiar sequences located in the 3′ UTR that provide SELENOP mRNA with stem-loop structures are called selenocysteine-insertion sequences (SECISs). In the presence of SECISs, the UGA codons in SELENOP mRNA are translated to Sec residues in a manner dependent on Sec translational machinery, including SECIS binding protein 2 (SBP2),1) although some of the UGAs might work as interrupting stop codons before the UAA.7) SECISs are common among selenoprotein-coding genes; however, having more than one is unique to SELENOP, suggesting the necessity of two but not more SECISs for the translation of multiple UGA codons to Sec.
The matured predominant isoform of human SeP protein consists of 359 amino acids (AA) after cleavage of the predicted signal peptide (AA 1–21). The protein migrates as multiple bands with molecular masses of approximately 50–60 kDa on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), presumably due to variations in glycosylation (conserved 2 N-linked- and one O-linked glycosylation7,11)); to different isoforms (per differential splicing and/or interrupting termination at one of the UGA codons, observed in rat SeP7)); and/or to proteolytic cleavage.12) As shown in Fig. 1, SeP consists of two functionally separable regions, the N-terminal catalytic region with the first Sec, and the C-terminal Sec/Cys-rich region. Two tandem histidine (His)-rich sequences and two potential cleavage sites for plasma serine (Ser) protease kallikrein are located between these two regions.12) In vitro experiments demonstrated that the N-terminal region exhibits peroxidase activity to reduce phospholipid hydroperoxides.12,13) As in the case for other Se-containing peroxidases GPxs, the enzymatic activity of SeP relies on the presence of a co-substrate, reportedly glutathione (GSH), thioredoxin (TRX), or other thiol-containing molecules with some hydroperoxides, for redox turnover of the catalytic Sec residue.13,14) However, neither the physiological substrates nor endogenous co-substrate(s) in vivo have been fully determined; in particular, the physiological concentration of the above candidates for co-substrates in plasma may be insufficient.
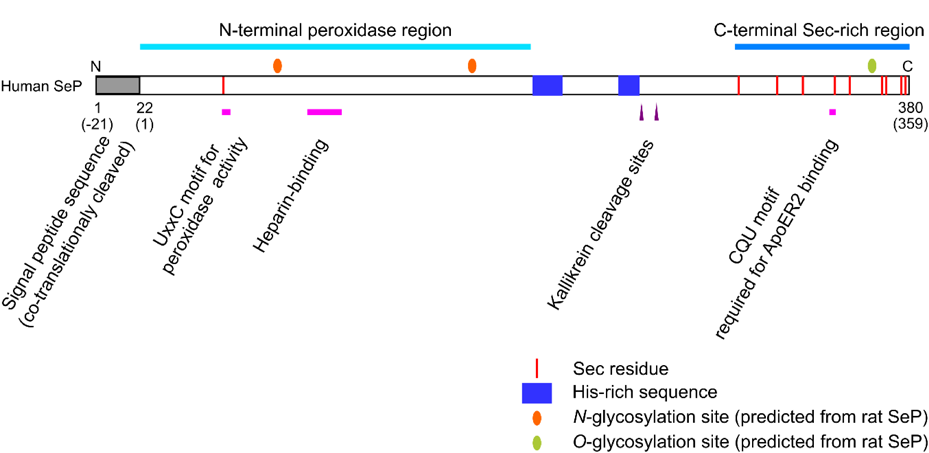
(Color figure can be accessed in the online version.)
The three-dimensional structure of SeP has not been solved heretofore; one possible reason is the difficulty in the exogenous overexpression of SeP in bacteria or in cultured cells because of the presence of multiple Sec residues in the polypeptide.
2.2. Regulation of SePIn literature, reported plasma concentrations of SeP in healthy subjects vary between 0.2–6 µg/mL, most likely because of divergent and yet unstandardized methods for quantification, leaving a possibility of geographical reasons for these variations related to selenium intake. If, assuming plasma SeP concentration is 5 µg/mL and SeP contains 6.3 Sec residues on average, according to our own quantification, then the plasma SeP level is equivalent to approximately 140 nM of SeP and 900 nM of Sec, respectively.
Orally administered 75Se-selenite to rats resulted in immediate and efficient incorporation of radioactivity to plasma SeP through the liver in a manner dependent on the portal vein, which delivers absorbed Se from gut to liver.15) A significant loss of plasma SeP was observed in mice with liver-specific homozygous deletion of Trsp, the Sec tRNA[Ser]Sec gene, which is necessary for selenoprotein synthesis, or of Selenop, by 75 and 90%, respectively.16,17) These results highlight an indispensable role of hepatocytes as the primary source of systemic plasma SeP, at least in rodents. By contrast, the expression of Selenop mRNA seems rather ubiquitous among tissues in rat, including the kidney, testis, lung, spleen, heart, and brain, although the liver had the highest level.18) In humans, the maximum SELENOP mRNA was found in liver, but with lesser expression levels in the small intestine, spleen, colon, gall bladder and other tissues.19) The biological significance of non-liver derived SeP remains controversial (also see section 2.3), but, especially under pathophysiological conditions, SeP is also reportedly produced in tissues other than liver20,21) (see later sections).
Cellular production of selenoproteins, in general, is increased upon Se uptake, and this induction likely involves multiple steps including transcriptional regulation, mRNA stabilization, potentiation of translational efficiency, etc.22) In addition to these common Se-driven mechanisms for selenoproteins, the production of SeP in the liver is regulated negatively by other pathways at the transcriptional level, including those evoked by cytokines such as IL1β, IFNγ, TNFα and TGFβ, or a transcription factor SREBP1, depending on cis-elements in the promoter region.23–26) In contrast, transcription factors FoxO1, FoxO3, PGC1α or HIF1α enhance the transcription of SELENOP.21,27,28) Additionally, SeP and the AMP activated protein kinase (AMPK)-FoxO1 pathway, a cellular energy sensor, regulate each other,29–32) implying an involvement of SeP in metabolic homeostasis and abnormalities. However, the entire mechanism underlying the regulation of plasma SeP concentration remains elusive, and must be scrutinized further for the epigenetic regulation, protein synthesis, maturation/glycosylation, and secretion of SeP, as well as its turnover in plasma, excretion and degradation. Moreover, although several post-translational modifications of SeP have been reported, including glycosylation and proteolytic cleavage in plasma, it is still unknown whether these are changed dynamically under physiological or pathophysiological conditions, whether they alter the biological activities of SeP, and whether they have biological significance.
2.3. SeP as a Se CarrierAn early observation showed that an intracardiac injection of 75Se-labeled SeP to a rat resulted in accumulation of the radioactivity in the kidney, testis and spleen, as well as the liver.4) Further, cells cultured with SeP-deficient serum became Se deficient, and supplementation of SeP to the Se-depleted cultured cells efficiently restored cellular glutathione peroxidase (GPx) activity.33,34) These facts provided an idea that SeP supplies Se to tissues. This “Se carrier” model was supported by subsequent studies in which the testicular Se content in Selenop−/− mice was sharply depressed, even in a Se-sufficient dietary condition, concomitant with male infertility, without exhibiting structural abnormalities in the seminiferous tubules.35,36) In addition, when these SeP-deficient mice were fed a Se-insufficient diet, the Se content in the brain and kidney were also decreased compared to control wild-type mice.35,36) Of these, the testis- and brain-associated phenotypes were recapitulated in knock-in mice which express a SeP mutant that lacks the C-terminal region, revealing that the Sec-rich sequence is the Se “shipper.”37) Meanwhile, a SeP knockout-dependent decrease in renal Se content, together with an increased urinary excretion of the element, is thought to reflect an involvement of SeP in proper maintenance of the systemic Se level.38) Nonetheless, these observations indicated that SeP works as a Se carrier to specific tissues, especially the testis, brain and kidney. However, the mechanism(s) that dictates tissue specificity for SeP-dependent Se transportation is(are) not fully clarified; one simple idea for this specificity is that the testis and brain require a continuous supply of SeP to maintain Se levels. Another explanation could be that there is(are) other form(s) of Se than SeP, possibly such as selenite or selenomethionine, in plasma that cannot reach the testis, but can reach other tissues, although slightly inefficiently for the brain. Cellular intake of non-SeP Se may require a cell-surface transporter(s), and these cells in the testis and brain may express to a lesser degree. Alternatively, given the presence of the blood–brain and blood–testis barriers,39,40) only Se in the form of SeP could efficiently pass through such boundaries for delivery. Interestingly, Schweizer et al. demonstrated that SeP can be synthesized in the brain, which contributes to intra-brain Se storage and prevention of neurological defects.41) It is therefore conceivable that SeP is involved in Se-maintenance in the testis as well.
To date, apolipoprotein E receptor 2 (ApoER2), megalin and low-density lipoprotein receptor-related protein 1 (LRP1), all of which are cell surface low density lipoprotein receptor (LDLR) family members,42) have been identified as SeP receptors in mouse testis and brain,43,44) kidney,45) and human myoblasts,46) respectively.
ApoER2 is best known as the receptor for reelin, an extracellular glycoprotein that is involved in early neuronal development and synaptic plasticity.47) ApoER2 knockout (Lrp8−/−) mice mimicked some of the phenotypes seen in Selenop−/− mice, i.e., depressed Se in the testis and brain, demonstrating an essential role for the ApoER2-SeP interaction in Se homeostasis in these organs, and (at least) dual receptor functions of ApoER2 as reelin- and SeP-receptors.43,44,48) The interaction between SeP and ApoER2 requires the C-terminal region of SeP, especially a conserved Cys/Sec-glutamine (Gln)-Cys/Sec motif (see Fig. 1), and the β-propeller domain of ApoER2.49) ApoER2 is expressed mainly in the brain, testis and thyroid,19) but also in a variety of tissues and in many cultured cell lines. It should be noted that ApoER2 has a diversity of isoforms with combinations of different functional domains, which may therefore differ in ligand-specificities, ligand-affinities and intracellular signaling. It is not yet fully understood which splicing variants of ApoER2 are expressed in different tissues/cells. Megalin is expressed almost exclusively in the kidney, specifically on the apical surface of renal proximal convoluted tubule cells, where it plays crucial roles in the glomerular filtrate function to recover proteins from urinary excretion, whereas the expression of LRP1 is reportedly ubiquitous. Molecular bases for SeP–megalin and SeP–LRP1 interactions (e.g. responsible regions in SeP, megalin or LRP1) are not yet specified, but the responsible region involved in SeP binding to megalin supposedly differs from that to ApoER2.45) Meanwhile, some of these LDLR members may cooperatively form a coreceptor for SeP, as suggested by data that the small interfering RNA (siRNA)-mediated depletion of either LRP1 or ApoER2 in myoblasts resulted in a significant deficiency in SeP uptake (see below).46)
In vitro cell culture experiments showed that SeP can bind to multiple cell lines, and that SeP is incorporated into cells in a receptor dependent manner, probably via endocytosis, and thereby enhances the cellular expression of other selenoproteins by providing a source of Se.46,50,51) The mechanism for Se recycling from SeP involves lysosomal SeP degradation, selenocysteine lyase (SCL)-dependent extrication of H2Se, and incorporation to tRNA[Ser]Sec, followed by newly synthesized selenoproteins.50,52) Of note, however, SeP-dependent Se delivery to cells in vitro should be carefully interpreted, given that mouse genetics clearly indicated predominant SeP-independent Se delivery to cells, as aforementioned.
Concomitant with the binding to and incorporation of SeP, LDLR proteins may elicit ligand-dependent intracellular signaling, for example, through the recruitment and activation of scaffold proteins and signaling proteins such as DAB and SFKs, as reported for ApoER2-reelin interaction.21) However, molecular bases for such SeP-dependent but Se-independent signaling have not yet been assessed in detail, and the difference in signaling among those evoked by SeP and other LDLR ligands, such as reelin, have not yet been established.
SeP is likely a housekeeping protein of which plasma concentration may not dynamically fluctuate in a physiological condition if Se intake is stable, although significant changes have been reported under pathophysiological conditions, such as cancer, metabolic syndrome, type 2 diabetes mellitus (T2DM), nonalcoholic fatty liver disease (NAFLD), and pulmonary arterial hypertension (PAH). Among these, recent studies suggest that changes in SeP are sometimes not merely consequences, but have potential as drivers, especially for T2DM and PAH, as noted below.
3.1. T2DM and SePT2DM is caused and/or promoted by a complex interplay of multiple disease-associated abnormalities, including high blood glucose, the acquisition of insulin resistance, and a temporal increase and subsequent decrease in blood insulin level. The connection between T2DM and an excess of Se was indicated by an epidemiological analysis in the US, where the average base-line blood Se levels is reportedly higher than in other regions. The study showed that high dose supplementation of Se unexpectedly increased the risk of developing the disease.53,54) Meanwhile, Misu and colleagues independently identified SeP as a T2DM-associated “hepatokine,” of which the mRNA levels in liver, as well as the protein levels in plasma, were elevated in diabetic patients compared to nondiabetic subjects, with significant correlations between SeP levels and the T2DM indicators such as blood glucose level and HbA1c.29) Noteworthy, the report clearly showed that excess plasma SeP induces insulin resistance in wild-type mice, whereas the siRNA-mediated depletion of SeP ameliorates insulin sensitivities in T2DM model mice. SeP caused cellular insulin resistance of hepatocytes and myocytes in vitro, detected by the suppression of IR autophosphorylation, reduction of the downstream AKT pathway responding to insulin, and a decline in insulin-evoked cellular glucose uptake. SeP-induced insulin resistance likely involves cellular glutathione, suggesting a potentially critical role for de novo syntheses of selenoenzymes that utilize glutathione as a co-substrate for redox-related reactions. However, the molecular mechanisms underlying this SeP-induced acquisition of insulin-resistance vary among tissues/cells; SeP caused insulin-resistance through the suppression of the AMPK pathway at least in part in hepatocytes, whereas SeP induced the insulin-resistance of myocytes independently of the AMPK pathway.29) Furthermore, we recently reported that excess plasma SeP causes not only insulin resistance, but also depletes insulin production by eliminating pancreatic β cells.51) Another observation illuminated an involvement of SeP in impaired angiogenesis, often seen in T2DM patients, showing the cellular acquisition of vascular endothelial growth factor (VEGF)-resistance by excess SeP.55) Together, these findings strongly indicate that overloaded SeP alone can cause and/or promote multiple aspects of T2DM, at least in mice. Of note, the expression of SeP is regulated positively by high glucose concentration29,55,56) and negatively by insulin.27–29) In addition, excess SeP causes depletion of blood insulin and elevation of blood glucose.29,51) Accordingly, SeP overexpression, along with T2DM symptoms, can be accelerated easily once there is an imbalance, even with a small fluctuation at the beginning (Fig. 2). Interestingly, metformin, one of the most established medicines for T2DM, suppresses SeP expression and secretion from hepatocytes,31,56) possibly providing another novel mode-of-action of this drug. Together, these studies have established SeP as an emerging central player in the initiation and/or progression of T2DM.
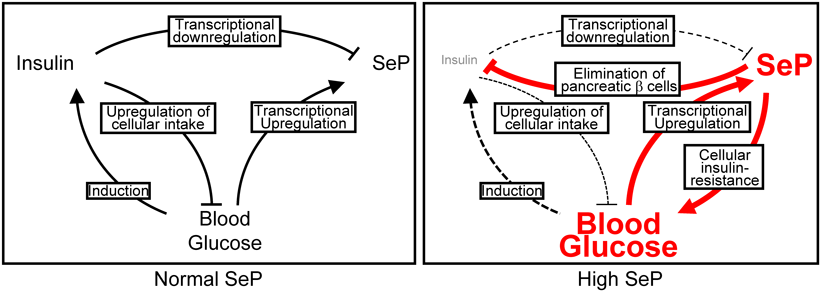
Plasma SeP level is regulated negatively by insulin-mediated signaling in hepatocytes in a normal condition. High blood glucose possibly induces excess plasma SeP, resulting in insulin resistance as well as the elimination of pancreatic β cells, thereby further elevating plasma SeP. (Color figure can be accessed in the online version.)
The correlation between SeP levels and T2DM symptoms has been assessed in several studies which have shown a stepwise increase of plasma SeP among normal, prediabetic and diabetic subjects,57) a correlation between plasma SeP and fasting plasma glucose, and an inverse correlation between plasma SeP and another T2DM marker, adiponectin,58) as well as a correlation between high plasma SeP and fasting blood glucose, although there was no correlation with HbA1c.59) In these studies, however, plasma SeP levels in control subjects were reported to be much lower than our own results, presumably reflecting different assay kits used for quantification, which are not necessarily standardized.
3.2. PAH and SePIn addition to T2DM, another example of pathophysiological roles for excess SeP was identified recently in PAH. Currently, PAH is thought to be an incurable disease associated with thickened pulmonary artery walls, due to the dysregulated proliferation of vesicular endothelial and/or smooth muscle cells, and thereby elevated pulmonary blood pressure, eventually resulting in right ventricular failure. Mechanisms for PAH have been vigorously investigated worldwide, and several models have been proposed thus far, including dysregulated growth factors and cytokines (e.g. TGFβ, PDGF and FGF), and hypoxia-induced vasomotion and vesicular remodeling.60) Using PAH patient-derived lung tissue and pulmonary artery smooth muscle cells (PASMCs), as well as Selenop−/− mice, Kikuchi and coworkers showed that 1) plasma SeP in PAH patients was elevated likely due to SeP overexpression in PASMCs, but not in the liver; 2) PAH-PASMCs secrete and receive SeP, as an autocrine signal, to promote their own proliferation and survival by activating the AKT, extracellular signal-regulated kinase (ERK)-mitogen-activated protein kinase (MAPK) and HIF1α pathways, probably through ApoER2; 3) SeP is induced in mice when exposed to hypoxia (10% O2) that causes the muscularization of pulmonary arteries and pulmonary hypertension, mimicking human PAH; and, 4) Selenop−/− mice were resistant to the induction of PAH-related symptoms by hypoxia compared to wild-type animals, whereas the exogenous expression of SeP in wild-type mice exacerbates the symptoms.21) Together, these demonstrated an essential role of SeP in PAH progression, suggesting SeP as a therapeutic target for this heretofore undruggable disease.
3.3. Other DiseasesSeveral reports have indicated positive correlations between high plasma SeP levels and obesity and/or metabolic syndromes, as well as nonalcoholic fatty liver disease,58,61–63) although this is still controversial, as others have reported an inverse correlation.59,64,65) Of note, however, these results were derived from using not necessarily the same method, suggesting the need for standardization of SeP quantification. Further efforts are needed to clarify the entire relationship between plasma SeP level and metabolism, as proposed previously.66)
In contrast to the examples described above associated with an increase in plasma SeP, decreases in plasma SeP level also relate to several human diseases. Low plasma SeP levels have been reported in patients with prostate cancer67,68) and colorectal cancer.69) Also, retrospective studies have suggested low plasma SeP levels as a risk of lung cancer in those of African ancestry70) and a risk of hepatocellular carcinoma.71) Low plasma SeP also correlated with the poor survival of renal cancer patients.72) Additionally, several single nucleotide polymorphisms (SNPs) in the SELENOP allele were reported to be associated with a higher risk of prostate cancer73) and colorectal adenoma.74) These reports suggest potential uses for plasma SeP level and SELENOP genotypes for the prediction, diagnosis and prognosis of cancer; yet, causal relationships as well as underlying mechanisms are poorly understood. Low SeP level is proposed to induce genomic instability, and to promote a protumorigenic inflammatory microenvironment, probably through the recruitment of cancer-associated M2 macrophages.20) Intriguingly, low plasma SeP is also reported in patients with inflammatory bowel disease,75) which is considered to be associated with colorectal and other carcinomas.76)
Additionally, decreases in plasma SeP are also reported in patients with strokes or other cardiovascular diseases,77,78) with septic shock or systemic inflammatory response syndrome,79) and with silicosis.80)
As described, many studies have indicated that SeP is involved, sometimes causally, in diseases. Because of the relative ease in obtaining blood/plasma samples from human subjects, there are emerging attempts to utilize patients’ plasma SeP level as one of the biomarkers for predictions or diagnoses/prognoses, possibly in combination with other markers. Intriguingly, a recent prospective study suggested that plasma SeP concentration can predict a patient’s insulin tolerance after 4 years, better than other indexes such as fasting blood glucose or HbA1c.81) For PAH patients, lower plasma SeP levels were significantly correlated with longer event-free survival.21) In addition, as noted above, given the association of lowered plasma SeP levels with human malignancies, SeP level could also be employed in early diagnoses and/or prognoses of cancer. A present technical obstacle, however, is that sensitivities and accuracies of currently available assay methods/kits vary significantly.82) Therefore, a standardized method is needed to achieve the use of plasma SeP as a biomarker in clinical situations.
4.2. Targeting SeP for Prophylaxis and TreatmentGiven that heterozygous deletion of the Selenop allele in mice does not produce phenotypes with serious conditions, such as male infertility and neuronal deficiencies, which were seen in animals with homozygous deletion,35,36) the suppression of plasma SeP, even to half the basal level, could be a potential therapeutic strategy to ameliorate excess SeP-driven disorders without causing toxicity. Several approaches may be taken to achieve the appropriate suppression of plasma SeP. Tajima-Shirasaki et al. reported that treatment with eicosapentaenoic acid (EPA), a component of ω-3 polyunsaturated fatty acids (PUFAs) found in fish oil, suppressed SeP expression in hepatocytes, although this has not been examined in animal experiments.26) Recent high-throughput screening identified sanguinarine, a plant alkaloid, as a strong suppressor of SELENOP mRNA expression in PAH-PASMCs that has been shown to prevent hypoxia-induced pulmonary hypertension in model mice.21) Finally, we reported a more direct approach, the injection of a SeP-neutralizing antibody to subjects, and indicated that this antibody-therapy ameliorated T2DM symptoms in model mice.51) It is still necessary to examine whether suppressing or neutralizing excess plasma SeP can benefit for prevention of or recovery from these diseases. Nevertheless, these approaches are promising for providing novel or alternative therapies, although they still need to be evaluated and modified for efficacy and safety, especially for human use.
We have listed, thus far, the involvement of SeP in human diseases and possible applications targeting SeP; however, especially from cell biological and biochemical points of view, there are still mysteries to be elucidated regarding the pathophysiological roles of SeP. One of these questions is why excess SeP causes such a variety of cellular reactions. In the context of T2DM, excess SeP results in insulin- or VEGF-resistance of hepatocytes and myocytes or of vascular endothelial cells, respectively, suppressing their mitogenic/survival signals such as the MAPK or AKT pathway.29,55) Contrarily, excess SeP promotes the proliferation and survival of smooth muscle cells in PAH, through aberrant activation of these pathways.21) Meanwhile, excess SeP induces pancreatic β cell death by an as yet unknown mechanism.51) From in vivo and in vitro studies, the biological activities of SeP can be dissected mainly as follows: A) peroxidase activity in plasma, B) Se supply activity, C) receptor-mediated intracellular signaling, and others (Fig. 3). Thus, the pathophysiological activities of excess SeP, as observed in T2DM and PAH for instance, would be attributed to a plethora of one or some of these activities. Among these, A) is thought to contribute to the homeostasis of extracellular oxidative status in a physiological situation, and excess SeP would therefore result in an oxidative stress-free or a hyper-reductive environment in which the effects on cells are totally unknown. For B), overloaded Se delivery to cells by excess SeP may cause the overexpression of a set of selenoproteins and/or the accumulation in cells of non-peptide forms of Se, possibly H2Se or selenite; the responses may also diverge among tissues/cells. Excess Se intake may also evoke an intracellular hyper-reductive condition through unregulated expression of redox-related selenoenzymes. Of note, however, in vitro cell culture experiments indicated that Se supply, measured by means of cellular GPx activity, saturates by treatment with much lower concentrations of SeP than in a physiological condition,33) suggesting no further increase in SeP-dependent Se supply on a transition from physiological to pathophysiological extracellular SeP levels. The activity described as C) might be more complicated because the expression pattern of receptors and downstream signaling proteins could vary among tissues/cells, thereby resulting in totally different cellular outputs, possibly including different Se-evoked expressing subsets of selenoproteins. The excessively reductive environment in/around cells caused by overloaded SeP and/or the SeP-evoked overexpression of selenoproteins is proposed to suppress “signaling ROS” required for activation of the AMPK pathway and for the expression of PGC-1α, a master regulator of mitochondrial biogenesis. Interestingly, such a SeP-caused hypo-oxidative stress condition in skeletal muscle was found to abolish an exercise-triggered metabolic adaptation of muscles,46) to which a coincidence of high blood SeP and obesity58,61–63) may be attributed.
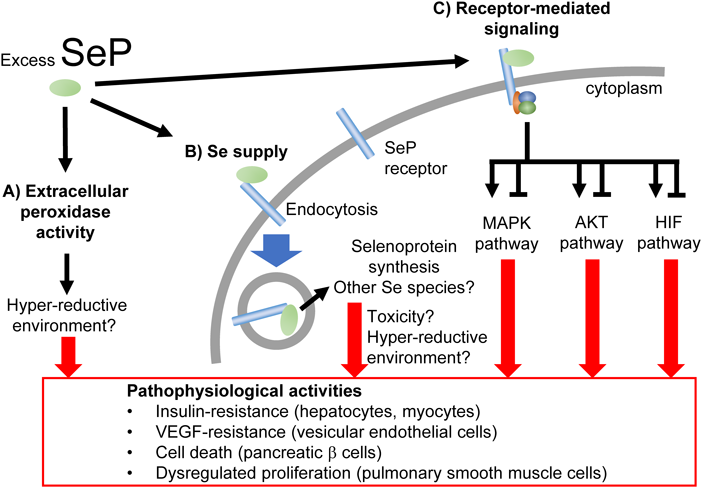
Excess SeP evokes cell/tissue context-dependent responses through its multiple activities. (Color figure can be accessed in the online version.)
Additionally, one could consider that SeP, as a receptor ligand, is relatively abundant in plasma even in healthy subjects (approx. 100 nM) when compared to known growth factors or cytokines (typically pM order), and that SeP-receptor binding should occur in a physiological condition, possibly at a semi-saturating level, based on the observed Se delivery efficacy of SeP to cultured cells.33) This begs the question, how does an excess of SeP, but not normal levels of SeP, cause pathological signaling? To answer, it will be helpful to know the dissociation constants of SeP and its receptors, the expression profiles of each receptor in different cells/tissues, and/or the signaling that is changed depending on extracellular SeP concentration, probably in a multi-phased manner. Nonetheless, the biological effects of SeP in pathophysiological conditions are apparently multi-faceted and complicated; therefore, it is quite important to clarify which of the SeP activities play(s) the major role in each disorder. Revealing these disease/cell/tissue context-dependent mechanisms will help us to understand the SeP-dependent biological significance, as well as help us to develop further effective pharmaceutical applications targeting SeP.
Accumulating evidence has indicated pivotal roles of SeP beyond the previous view that the protein simply reflects Se intake and contributes to systemic Se supply; instead, SeP behaves as something of a villain when aberrantly overexpressed. Especially, recent discoveries of the causal and/or promoting effect of excess plasma SeP in several diseases has revealed a new aspect of SeP: that of an emerging therapeutic target. Currently, however, medical/pharmaceutical applications targeting SeP is still far from the drug development stage. To this end, further studies are needed at all levels of molecular/cellular biology, drug discovery, animal experiments, human epidemiology, and clinical research.
This work was supported by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (19K21238 to R.T.; 17H03821 and 19K22480 to Y.S.) and by the Takeda Science Foundation (to R.T.).
The authors declare no conflict of interest.