Abstract
Tyrosine kinase 2 (Tyk2) is a member of the Janus family of protein tyrosine kinases (Jaks). Tyk2 associates with interferon (IFN)-α, IFN-β, interleukin (IL)-6, IL-10, IL-12, and IL-23 receptors and mediates their downstream signaling pathways. Based on our data using Tyk2-deficient mice and cells, Tyk2 plays crucial roles in the differentiation, maintenance, and function of T helper 1 (Th1) and Th17 cells, and its dysregulation may promote autoimmune and/or inflammatory diseases. IFN-α-induced growth inhibition of B lymphocyte progenitors is dependent on Tyk2-mediated signals to regulate death-associated protein (Daxx) nuclear localization and Daxx-promyelocytic leukemia protein interactions. Tyk2-deficient mice show impaired constitutive production of type I IFNs by macrophages under steady-state conditions. When heat-killed Cutibacterium acnes is injected intraperitoneally, Tyk2-deficient mice show less granuloma formation through enhanced prostaglandin E2 and protein kinase A activities, leading to high IL-10 production by macrophages. Thus, Tyk2 is widely involved in the immune and inflammatory response at multiple events; therefore, Tyk2 is likely to be a suitable target for treating patients with autoimmune and/or chronic inflammatory diseases. Clinical trials of Tyk2 inhibitors have shown higher response rates and improved tolerability in the treatment of patients with psoriasis and inflammatory bowel diseases. Taken together, Tyk2 inhibition has great potential for clinical application in the management of a variety of diseases.
1. INTRODUCTION
Tyrosine kinase 2 (Tyk2) was originally identified as a protein with biological activity pertaining to recovering responsiveness against type I interferon (IFN) in an IFN-unresponsive human cell line generated by saturation mutagenesis.1) Experiments using Tyk2-deficient mice and cells have demonstrated that Tyk2-mediated signals are involved in both innate and acquired immune responses via an increase in the numbers and functions of T helper 1 (Th1) and Th17 cells, as well as contributing to constitutive production of type I IFNs.2–5) In addition, Tyk2-mediated signals play an essential role in down-regulating the in vivo production of interleukin (IL)-10.6,7) Thus, Tyk2 widely mediates signals to control immune and inflammatory responses in multiple steps.
Progress in understanding the pathogenesis and molecular abnormalities underlying autoimmune and/or inflammatory diseases has expanded the scope of novel therapeutic strategies. Cytokines themselves, as well as their induced signals, may be suitable targets. In addition to applications of monoclonal antibodies against cytokines, low-molecular weight compounds that inhibit signaling molecules have also been identified as an effective strategy, and some are clinically approved for the treatment of patients with autoimmune diseases. In particular, the Janus family of protein tyrosine kinases (Jaks) is a promising target.8)
Herein, we review the data of our experiments on Tyk2-deficient mice and the molecular mechanisms underlying Tyk2 involvement in immune and inflammatory systems, as well as the current situation, to develop novel Tyk2 inhibitors.
2. STRUCTURE OF TYK2
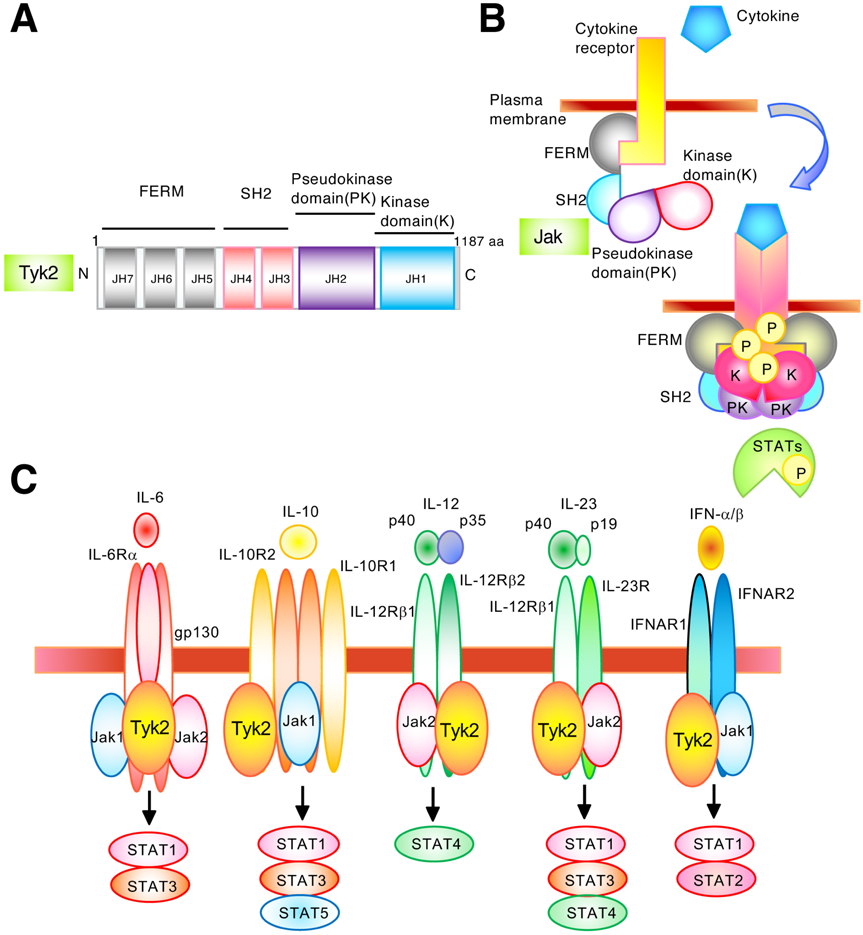
Tyk2 is a member of the Jak family, with an apparent molecular weight of approximately 120–140 kDa.1) An alternative nomenclature for the putative domains is given as a series of Janus homology (JH) domains.9) From the information of amino acid sequence, Tyk2 commonly contains N-terminal four-point-one, ezrin, radixin, moesin (FERM) homology, Src homology 2 (SH2), pseudokinase, and kinase domains1,9,10) (Fig. 1A). The FERM domain binds to cytokine receptors and the SH2 domain associates with phosphorylated tyrosine residues. The tyrosine kinase domain (JH1) increases its catalytic activity by trans- and/or auto-phosphorylation of the activation loop as a result of conformational changes at ligand-bound receptors. The pseudokinase domain (JH2) is catalytically inactive but interacts with the kinase domain, leading to negative regulation of its catalytic activity.
3. JAK–SIGNAL TRANSDUCER AND ACTIVATOR OF TRANSCRIPTION (STAT) PATHWAY
Cytokine receptors are constitutively associated with inactive Jaks. The binding of cytokines to their receptors induces a conformational change in the receptor complex, altering the alignment of receptor-associated Jaks, leading to their phosphorylation and activation9,11–13) (Fig. 1B). The activated Jaks further phosphorylate tyrosine residues in the cytoplasmic tail of receptors, which serve as docking sites for the members of STAT family. Cytoplasmic STATs then bind to the phosphorylated receptors and become substrates for Jaks. Phosphorylated STATs form dimers, followed by dissociation from receptors and their accumulation in the nucleus, where they promote cytokine-responsive gene expression. Thus, the Jak–STAT signaling pathway is widely utilized by members of the cytokine receptor superfamily to induce multiple cellular events.
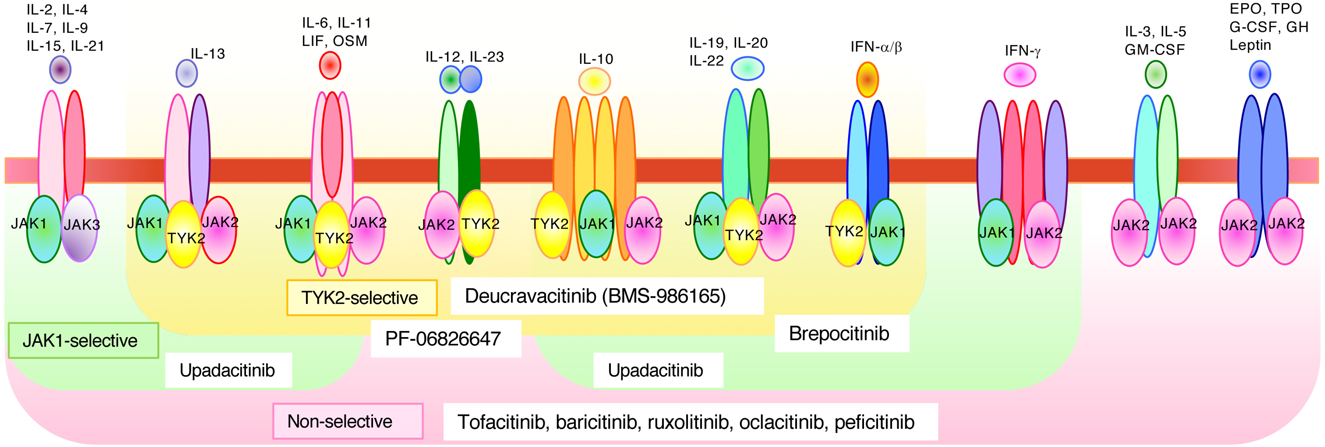
Jak1 and Jak3 associate with a common γ chain, which is shared among receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Jak1 is also essential for other cytokine families that use a shared receptor subunit gp130, including receptors for IL-6, IL-11, oncostatin M, leukemia inhibitory factor, granulocyte colony-stimulating factor (G-CSF), and IFNs.8,9) Jak2 associates with receptors for hormone-like cytokines, such as growth hormone, prolactin, erythropoietin, and thrombopoietin, as well as cytokines that mediate signals through an IL-3 receptor subunit, including IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF).8,9) Jak2 is also essential for few IFNs and cytokine receptors that consist of a gp130 subunit. Tyk2 associates with receptor subunits, such as IL-12Rβ1, IL-10R2, IL-23R, IFNAR1, and gp1308,9) (Fig. 1C).
Jak1-deficient mice perinatally die and lack the development and function of lymphocytes.14) Jak2-deficient mice show embryonic lethality due to failure of definitive erythropoiesis.15) Jak3-deficient mice show defects in immune cell development and function, and Jak3-deficiency is detected in patients with autosomal recessive severe combined immunodeficiency.16,17) Tyk2-deficient mice are viable but susceptible to viral infection and exhibit insufficient responses to lipopolysaccharide (LPS).2,3,18,19)
4. EFFECTS OF TYK2 ON CYTOKINE SIGNALING
Tyk2 associates with certain subunits of cytokine receptor complexes, such as IFNAR1, IL-12Rβ1, IL-10R2, and IL-23R8,9) (Fig. 1C). Tyk2 is also associated with the gp130 receptor subunit.8,9) IL-12 recognizes receptors carrying IL-12Rβ1 and promotes cell-mediated immunity to protect the host from infection.20) IL-22 recognizes receptors carrying IL-10R2 and plays an essential role in wound healing, tissue barrier function, epithelial repair, and homeostasis.21) IL-23 recognizes receptors that contain IL-12Rβ1 and induces inflammatory responses. Moreover, IFN-α and IFN-β recognize receptors that contain IFNAR1.8,9)
Although Tyk2-deficient B lymphocyte progenitors show impaired growth inhibition induced by IFN-α,22) Tyk2 is known to only have restricted effects on the IFN-α signaling pathway.2,3) In contrast, most IL-12-induced cellular functions completely disappear because of Tyk2-deficiency.2,3) Tyk2-deficient mice show less immunity and inflammatory phenotypes in several murine experimental models, including arthritis and colitis models.5,23) Importantly, patients with autosomal recessive hyper-immunoglobulin E (IgE) syndrome, who have a homozygous Tyk2 gene mutation to promote the loss of mature Tyk2 proteins, experience high serum IgE levels, recurring skin abscesses, and repeated pneumonia.24) Therefore, Tyk2-mediated signaling contributes to both the innate and acquired immune systems.
5. INVOLVEMENT OF TYK2 IN THE GENERATION AND FUNCTION OF TH1 AND TH17 CELLS
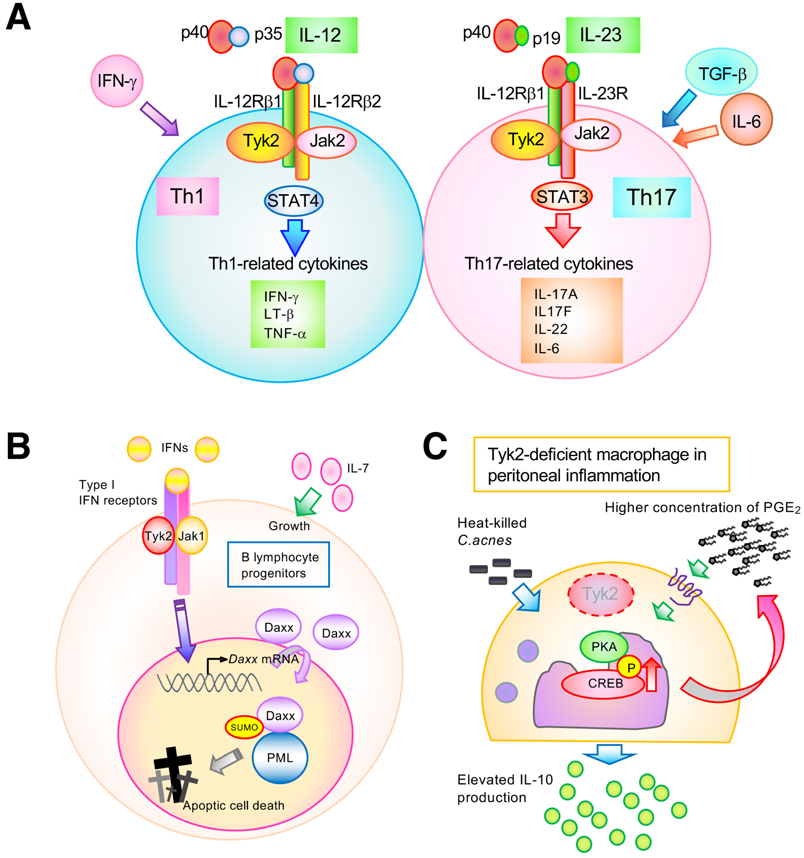
The binding of IL-12 to its receptors, which are associated with Tyk2 and Jak2, mainly activates the STAT4 transcription factor20) (Fig. 2A). Activated STAT4 cooperates with the signals from T cell receptor (TCR) to express T-bet, which is a master transcriptional factor that promotes the differentiation of naïve CD4+ T cells into Th1-type cells. Th1 cells are central players in inducing cell-mediated immune responses to defend against viral or bacterial pathogens.25) In addition, Th1 cells produce IFN-γ, IL-2, IL-10, and tumor necrosis factor (TNF)-α that participate in macrophage activation, cytotoxic T cell generation, and nitric oxide production. Binding of IL-23 to its receptor, which is associated with Tyk2, mediates signals for the proliferation, survival, and functional maturation of Th17 cells, while TGF-β and IL-6 are largely involved in Th17 cell differentiation (Fig. 2A). Th17 cells produce IL-17, IL-21, and IL-22, which induce inflammatory responses to eliminate microbial pathogens. However, prolonged and excessive activation of Th17 cells is believed to cause autoimmune and/or inflammatory diseases in humans.26) Thus, Tyk2 is highly involved in immune and inflammatory processes by positively regulating both IL-12/Th1 and IL-23/Th17 axis. In addition, dysregulation of Tyk2 can cause confusion in the immune system.5)
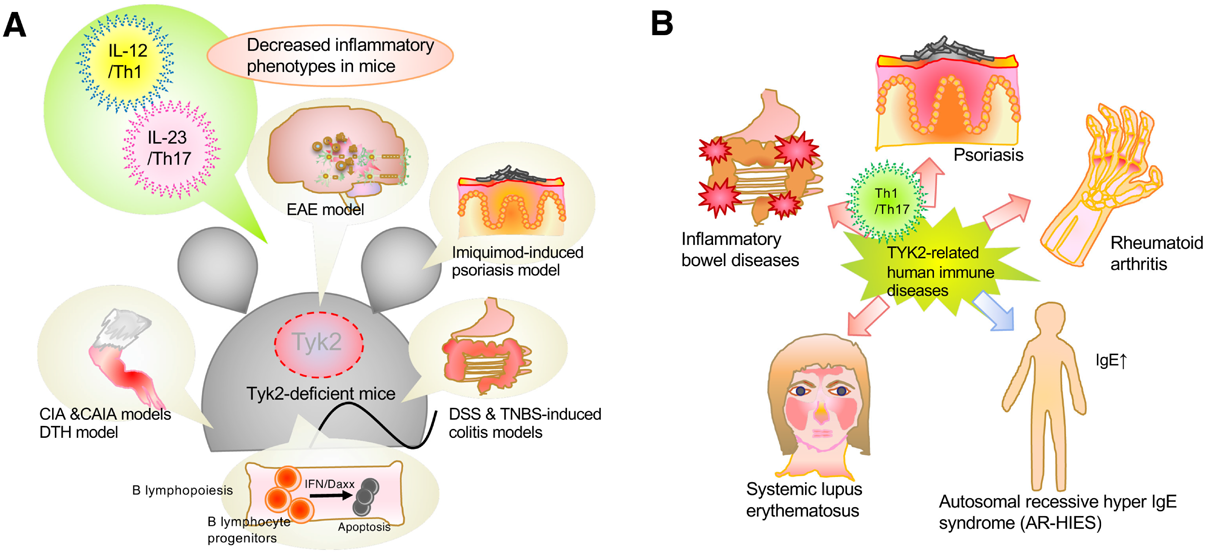
Using Tyk2-deficient mice or cells, we have reported the contribution of Tyk2 to pathological immune and/or inflammatory processes5,6,19,23) (Fig. 3A). Tyk2-deficient dendritic cells cannot produce IL-12 or IL-23 in response to cytosine-phosphate-guanine (CpG) oligodeoxynucleotides, resulting in the failure to promote differentiation of CD4-positive cells into Th1 cells.26,27) In both collagen-induced and anti-type II collagen antibody-induced experimental arthritis models, Tyk2-deficient mice showed significantly low susceptibility to arthritis.23) In an experimental autoimmune encephalomyelitis model, Tyk2-deficient mice showed lower clinical scores of symptoms and fewer lymphocytes that invaded the central nervous system.28,29) In both dextran sulfate sodium-induced and 2,4,6-trinitrobenzene sulfonic acid-induced colitis experimental models, Tyk2-deficient mice showed slower and lesser disease development, diarrhea, and body weight loss, compared with those by wild-type (WT) mice.5) In an experimental model of skin inflammation induced by imiquimod, a ligand for TLR7, Tyk2-deficient mice induced less severe epidermal hyperplasia, parakeratosis, and inflammatory cell invasion.5,30) Involvement of Th1 and Th17 cells in these experimental models are summarized in Fig. 3.
Therefore, Tyk2-mediated signals participate in host defense by positively regulating the generation and function of both Th1 and Th17 cells. In addition, Tyk2-deficiency reduces autoimmune and/or inflammatory phenotypes in murine experimental models (Fig. 3A).
6. REQUIREMENT OF TYK2 IN IFN SIGNALING
IFN-α potently promotes antiviral and antiproliferative activities. Stimulation of target cells with IFN-α selectively activates Tyk2 and Jak1, resulting in the induction of Stat1 and Stat2 phosphorylation.9) Although Jak1-deficient cells fail to respond to IFN-α, Tyk2-deficient cells are preferentially resistant to the suppression of B lymphocyte growth by IFN-α14,22) (Fig. 3A). In addition, Stat1-deficient mice completely fail to respond to IFN-α,31) and Stat2-deficient mice showed high susceptibility to viral infection.32)
The colony-forming unit in response to IL-7 (CFU-IL-7) is a useful tool for analyzing the growth capacity of B lymphocyte progenitors.33) Since no difference in CFU-IL-7 colony formation of bone marrow cells between wild-type and Tyk2-deficient mice was observed, IL-7-responding B lymphocyte progenitors were similarly generated independently of Tyk2 under steady-state conditions. However, IFN-α failed to reduce colony numbers of CFU-IL-7 in Tyk2-deficient bone marrow cells, while CFU-IL-7 colony formation in wild-type bone marrow cells was dramatically suppressed in the presence of IFN-α. In addition, the IFN-α-induced up-regulation and nuclear translocation of Daxx are completely abrogated in the absence of Tyk2. Regarding its possible mechanisms, Tyk2 is likely to be required for the elevation and nuclear translocation of death-associated protein (Daxx) induced by IFN-α22) (Fig. 2B). Daxx binds to Fas, resulting in the activation of a signaling cascade for apoptotic cell death.34) Sumoylation of Daxx is an essential event for its nuclear accumulation and binding to promyelocytic leukemia protein (PML).35) Importantly, IFN-α enhanced sumoylation and nuclear accumulation of Daxx in cells. Therefore, a sumoylation-defective Daxx K630/631 A mutant (Daxx KA) localizes to the cytoplasm, whereas wild-type Daxx localizes to the nucleus. Notably, the murine pro-B cell line Ba/F3 acquires resistance against IFN-α-induced growth inhibition when Daxx KA is overexpressed. In this case, the intracellular localization of Daxx KA was altered from the cytoplasm to the nucleus after treatment with leptomycin B, which is an exportin inhibitor. In parallel, leptomycin B treatment recovered IFN-α-induced growth inhibition in Ba/F3 cells expressing Daxx KA. Moreover, PML overexpression recovered the recruitment of Daxx KA into the PML oncogenic domains (PODs), which are subnuclear structures. Importantly, a Daxx-small ubiquitin-related modifier (SUMO) fusion protein provides increased Daxx nuclear localization and high cell growth inhibition. Thus, growth inhibition of B lymphocyte progenitors by IFN-α requires Daxx nuclear localization via its sumoylation and proper interactions between Daxx and PML. Usually, Daxx binds to PML and forms PODs. IFN-α treatment induces sumoylation of Daxx and PODs formation in cells. However, IFN-α treatment with Tyk2-deficinet B lymphocytes failed to form PODs, suggesting that Daxx-PML signaling is transmitted through Tyk2. Thus, Tyk2 selectively mediates signals to control Daxx-SUMO-PML interactions during IFN-α-induced growth inhibition of B lymphocyte progenitors.
7. INVOLVEMENT OF TYK2 IN CONSTITUTIVELY PRODUCED TYPE I IFNS
Constitutively produced type I IFNs, although the small in amount, regulate daily cell functions in an autocrine or paracrine manner and are needed for immediate and maximal immune responses against bacterial components.4) Tyk2-mediated signaling is involved in the constitutive production of IFN-α by macrophages.18) In Tyk2-deficient macrophages, basal and LPS-induced production of IFN-α is evidently impaired. In addition, type I IFN-related gene expression is significantly low in Tyk2-deficient macrophages, particularly under steady-state conditions.18)
Therefore, Tyk2 has certain effects on macrophage activation, leading to autocrine and/or paracrine production of type I IFNs. Tyk2 is essential for the daily production of type I IFNs to maximally enable the immune system in vivo when needed.
8. INVOLVEMENT OF TYK2 IN THE REGULATION OF IL-10 PRODUCTION
Intraperitoneal injection of heat-killed Cutibacterium acnes (formerly Propionibacterium acnes) produces obvious granulomas and causes neutrophil infiltration into the peritoneal cavity.6,7,36) Tyk2-deficient mice injected with C. acnes showed significantly fewer infiltrated neutrophils, lower pro-inflammatory cytokines, and higher IL-10 concentration in the peritoneal cavity than that observed in WT mice6,7) (Fig. 2C). Production of IL-10, which strongly suppresses inflammation, is believed to require autocrine type I IFN signaling. Although Tyk2 is involved in IFN production and signaling as mentioned above, pretreatment of WT mice with either anti-IFNAR1 or anti-IFN-γ antibodies cannot inhibit peritoneal inflammation induced by C. acnes injection. However, C. acnes-induced peritoneal inflammation was clearly reduced to a level comparable to that of uninjected mice by pretreatment of WT mice with a neutralizing antibody against the IL-10 receptor. Thus, the high production of IL-10 is responsible for the inflammation-suppressive phenotype in C. acnes-injected Tyk2-deficient mice, and Tyk2 seems to positively regulate the production of IL-10 in an autocrine IFN-independent manner.
IL-10-producing cells in peritoneal F4/80-positive macrophages were increased in Tyk2-deficient mice compared with those in WT mice.6,7) The number of IL-10-producing cells in the peritoneal F4/80-negative or B220-positive population did not change between Tyk2-deficient and WT mice. Thus, peritoneal macrophages mainly produce IL-10 in Tyk2-deficient mice after intraperitoneal injection of C. acnes. IL-10 production by macrophages has been reported to be upregulated by prostaglandin E2 (PGE2) signaling by enhancing the activity of protein kinase A (PKA).37,38) Moreover, C. acnes-induced IL-10 production by peritoneal macrophages was inhibited by the addition of diclofenac, an inhibitor of cyclooxygenases, which mediates the production of prostaglandins. Pretreatment with a specific PKA inhibitor, namely H-89, impeded C. acnes-induced IL-10 production by peritoneal cells. Importantly, the peritoneal lavage of Tyk2-deficient mice under steady-state conditions contained higher concentration of PGE2 than WT mice. Phosphorylation of cAMP response element binding protein (CREB), a hallmark of PKA activation, is induced by C. acnes stimulation alone and is further elevated by the addition of ectopic PGE2. Tyk2-deficient bone marrow-derived macrophages show elevated CREB phosphorylation in response to stimuli with C. acnes alone or in combination with C. acnes and PGE2.
In C. acnes-induced peritoneal inflammation, Tyk2-deficiency is likely to skew macrophage potential toward an anti-inflammatory state. Thus, the elevated IL-10 production observed in the peritoneal cavity of Tyk2-deficient mice may be attributed to the immunosuppressive microenvironment established by spontaneously high levels of PGE2–PKA activity. In other words, Tyk2 downregulates the PGE2–PKA–IL-10 pathway, resulting in a pro-inflammatory phenotype during inflammation caused by the intraperitoneal injection of C. acnes.
9. THERAPEUTIC ADVANTAGE OF TYK2 INHIBITORS
Cytokine-induced intercellular signaling is essential for the activation and regulation of the immune system, including host defense. Excessive and prolonged cytokine signaling sometimes becomes the major cause for the onset and development of autoimmune diseases; therefore, pharmacological inhibitors that target signaling pathways have been developed and approved to treat patients with several autoimmune and inflammatory diseases. Notably, the manufacturing of low-molecular-weight Jak inhibitors is relatively easier and cheaper than that of biologics. Furthermore, these inhibitors are available for oral consumption, which is a major advantage over biological agents. Currently, several Jak inhibitors, including tofacitinib, baricitinib, ruxolitinib, upadacitinib, oclacitinib, and peficitinib, have been approved by the Food and Drug Administration8) (Fig. 4). These compounds mainly bind to the ATP site of the catalytically active JH1 domain; therefore, they demonstrate inhibitory effects on multiple Jaks without high specificity. Currently, some Jak inhibitors have revealed clinical utility in rheumatoid arthritis, psoriasis, inflammatory bowel disease, myeloproliferative neoplasms, and graft-versus-host disease.8) Importantly, baricitinib has also been approved for the treatment of coronavirus disease 2019 (COVID-19) patients with moderate to severe symptoms.39) In the case of Tyk2-selective inhibitors,40–44) deucravacitinib (BMS-986165) was constructed to recognize the pseudokinase JH2 domain of Tyk2 and cause allosteric inhibition. This oral Tyk2-specific inhibitor was investigated in a phase 2 trial to treat patients with psoriasis. The other Tyk2 inhibitors were brepocitinib and PF-06826647. Brepocitinib recognizes the catalytic domains of Tyk2 and Jak1, and inhibits their kinase activity. Clinical trials of this oral inhibitory compound are now being conducted in patients with psoriasis and inflammatory bowel diseases. PF-06826647 is an oral inhibitor of Tyk2 and Jak2, and clinical trials of this compound are in progress for patients with psoriasis. Notably, the response rate was significantly higher in the Tyk2 inhibitor group than that observed in the placebo group. Additionally, the adverse effects of these Tyk2 inhibitors are clinically tolerable. Notably, Tyk2-deficient mice were viable and did not show severe phenotypes when compared to other Jak-deficient mice. Therefore, Tyk2 inhibitors will provide a more effective and safer strategy to treat patients with immune/inflammatory diseases compared to the currently available biologics.
10. CONCLUSION
Experiments using Tyk2-deficient mice or cells have clarified the involvement of Tyk2-mediated signaling in the immune and inflammatory systems. Tyk2 is essential for the differentiation, maintenance, and appropriate functions of Th1 and/or Th17 cells,5) which further promote immune and inflammatory responses that protect the host from infections. IFN-α-induced Tyk2 activation preferentially inhibits the growth of B lymphocyte progenitors through Daxx nuclear localization and proper interactions between Daxx and PML.22,35) Tyk2 is involved in the constitutive in vivo production of type I IFNs and thus facilitates a rapid response against invading pathogens. During peritoneal inflammation induced by C. acnes injection, Tyk2-mediated signals are required to suppress the in vivo production of IL-10 by regulating the PGE2–PKA pathway.6) Thus, as described in this review, Tyk2 regulates multiple cellular events during immune and/or inflammatory responses. Since Tyk2 is a key mediator in the immune system, Tyk2-deficient mice show significantly less immune/inflammatory phenotypes in several murine experimental models5,6,19,23,28–30) (Fig. 3A).
Genome-wide association studies have connected the Tyk2 loci with several autoimmune diseases, including rheumatoid arthritis, psoriasis, systemic lupus erythematosus, and inflammatory bowel diseases.45) A Tyk2 variant encoding a proline to alanine amino acid substitution at 1104 position, which is inactive, protects against several autoimmune diseases.46) Patients with variants of the Tyk2 coding sequence showed impaired IL-12, IL-23, and type I IFN signals. These findings confirm that Tyk2-mediated signals are critical for eliminating invasive foreign pathogens and that dysregulation of Tyk2 expression/function is involved in the onset and development of autoimmune diseases (Fig. 3B), and that ustekinumab, a monoclonal antibody against the p40 subunit shared by IL-23 and IL-12, has been approved for the treatment of patients with psoriasis and inflammatory bowel diseases.8) Antibodies against the IL-23-specific p19 subunit, such as guselkumab, tidrakizumab, and risankizumab, have been approved for the treatment of patients with psoriasis.8) The pharmacological profile of Tyk2 inhibitors may provide an opportunity to treat several autoimmune diseases with oral therapy.
As Tyk2 is involved in the signaling pathway induced by type I IFNs, IL-12, and IL-23, and dysregulation of Tyk2 function is highly responsible for the onset and development of autoimmune and inflammatory diseases, novel Tyk2 inhibitors are expected to have a significant clinical impact. Most commercially available Jak inhibitors bind to the active sites of their catalytic domains.8) However, a Tyk2-specific inhibitor such as deucracitinib can be designed to bind to its regulatory domain.40–44) Clinical trials of these inhibitors have shown satisfactory safety and efficacy in the treatment of patients with psoriasis and inflammatory bowel diseases. As described in this review, Tyk2 inhibitors can be used in a much wider clinical field of autoimmune or inflammatory diseases. Further investigation will provide useful information regarding the management of diseases using Tyk2 inhibitors.
Acknowledgments
This study was supported in part by a Grant-in-Aid for Scientific Research 19H03364 (T.M.) and 20K07010 (R. M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell, 70, 313–322 (1992).
- 2) Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, Yamashita M, Numata A, Takase K, Kobayashi S, Shibata S, Asano Y, Gondo H, Sekiguchi K, Nakayama K, Nakayama T, Okamura T, Okamura S, Niho Y, Nakayama K. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity, 13, 561–571 (2000).
- 3) Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, McCoy B, Bogdan C, Decker T, Brem G, Pfeffer K, Müller M. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity, 13, 549–560 (2000).
- 4) Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity, 36, 166–174 (2012).
- 5) Ishizaki M, Akimoto T, Muromoto R, Yokoyama M, Ohshiro Y, Sekine Y, Maeda H, Shimoda K, Oritani K, Matsuda T. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J. Immunol., 187, 181–189 (2011).
- 6) Hirashima K, Muromoto R, Minoguchi H, Matsumoto T, Kitai Y, Kashiwakura JI, Shimoda K, Oritani K, Matsuda T. The mechanism of Tyk2 deficiency-induced immunosuppression in mice involves robust IL-10 production in macrophages. Cytokine, 130, 155077 (2020).
- 7) Muromoto R, Oritani K, Matsuda T. Tyk2-mediated homeostatic control by regulating the PGE2-PKA-IL-10 axis. AIMS Allergy Immunol., 5, 175–183 (2021).
- 8) Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov., 16, 843–862 (2017).
- 9) Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Immunity, 36, 503–514 (2012).
- 10) Wallweber HJ, Tam C, Franke Y, Starovasnik MA, Lupardus PJ. Structural basis of recognition of interferon-α receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol., 21, 443–448 (2014).
- 11) Ihle JN. Cytokine receptor signalling. Nature, 377, 591–594 (1995).
- 12) Darnell JE Jr. STATs and gene regulation. Science, 277, 1630–1635 (1997).
- 13) O’Shea JJ, Ma A, Lipsky P. Cytokines and autoimmunity. Nat. Rev. Immunol., 2, 37–45 (2002).
- 14) Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, King KL, Sheehan KC, Yin L, Pennica D, Johnson EM Jr, Schreiber RD. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell, 93, 373–383 (1998).
- 15) Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, Grosveld G, Ihle JN. Jak2 is essential for signaling through a variety of cytokine receptors. Cell, 93, 385–395 (1998).
- 16) Nosaka T, van Deursen JM, Tripp RA, Thierfelder WE, Witthuhn BA, McMickle AP, Doherty PC, Grosveld GC, Ihle JN. Defective lymphoid development in mice lacking Jak3. Science, 270, 800–802 (1995).
- 17) Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O’Shea JJ, Vezzoni P, Notarangelo LD. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature, 377, 65–68 (1995).
- 18) Karaghiosoff M, Steinborn R, Kovarik P, Kriegshäuser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, Levy D, Decker T, Müller M. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol., 4, 471–477 (2003).
- 19) Kamezaki K, Shimoda K, Numata A, Matsuda T, Nakayama K, Harada M. The role of Tyk2, Stat1 and Stat4 in LPS-induced endotoxin signals. Int. Immunol., 16, 1173–1179 (2004).
- 20) Tait Wojno ED, Hunter CA, Stumhofer JS. The immunobiology of the interleukin-12 family: room for discovery. Immunity, 50, 851–870 (2019).
- 21) Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22–IL-22R1 system. Nat. Rev. Drug Discov., 13, 21–38 (2014).
- 22) Shimoda K, Kamesaki K, Numata A, Aoki K, Matsuda T, Oritani K, Tamiya S, Kato K, Takase K, Imamura R, Yamamoto T, Miyamoto T, Nagafuji K, Gondo H, Nagafuchi S, Nakayama K, Harada M. Cutting edge: tyk2 is required for the induction and nuclear translocation of Daxx which regulates IFN-alpha-induced suppression of B lymphocyte formation. J. Immunol., 169, 4707–4711 (2002).
- 23) Ishizaki M, Muromoto R, Akimoto T, Ohshiro Y, Takahashi M, Sekine Y, Maeda H, Shimoda K, Oritani K, Matsuda T. Tyk2 deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Int. Immunol., 23, 575–582 (2011).
- 24) Minegishi Y, Saito M, Morio T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity, 25, 745–755 (2006).
- 25) Swain SL, McKinstry K, Strutt T. Expanding roles for CD4+ T cells in immunity to viruses. Nat. Rev. Immunol., 12, 136–148 (2012).
- 26) Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu. Rev. Immunol., 27, 485–517 (2009).
- 27) Tokumasa N, Suto A, Kagami S, Furuta S, Hirose K, Watanabe N, Saito Y, Shimoda K, Iwamoto I, Nakajima H. Expression of Tyk2 in dendritic cells is required for IL-12, IL-23, and IFN-gamma production and the induction of Th1 cell differentiation. Blood, 110, 553–560 (2007).
- 28) Oyamada A, Ikebe H, Itsumi M, Saiwai H, Okada S, Shimoda K, Iwakura Y, Nakayama KI, Iwamoto Y, Yoshikai Y, Yamada H. Tyrosine kinase 2 plays critical roles in the pathogenic CD4 T cell responses for the development of experimental autoimmune encephalomyelitis. J. Immunol., 183, 7539–7546 (2009).
- 29) Spach KM, Noubade R, McElvany B, Hickey WF, Blankenhorn EP, Teuscher C. A single nucleotide polymorphism in Tyk2 controls susceptibility to experimental allergic encephalomyelitis. J. Immunol., 182, 7776–7783 (2009).
- 30) Ishizaki M, Muromoto R, Akimoto T, Sekine Y, Kon S, Diwan M, Maeda H, Togi S, Shimoda K, Oritani K, Matsuda T. Tyk2 is a therapeutic target for psoriasis-like skin inflammation. Int. Immunol., 26, 257–267 (2014).
- 31) Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell, 84, 443–450 (1996).
- 32) Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity, 13, 795–804 (2000).
- 33) Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, Mosley B, March CJ, Urdal D, Gillis S, Cosman D, Goodwin RG. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature, 333, 571–573 (1988).
- 34) Yang X, Khosravi-Far R, Chang HY, Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell, 89, 1067–1076 (1997).
- 35) Muromoto R, Ishida M, Sugiyama K, Sekine Y, Oritani K, Shimoda K, Matsuda T. Sumoylation of Daxx regulates IFN-induced growth suppression of B lymphocytes and the hormone receptor-mediated transactivation. J. Immunol., 177, 1160–1170 (2006).
- 36) Tanaka T, Yamamoto Y, Muromoto R, Ikeda O, Sekine Y, Grusby MJ, Kaisho T, Matsuda T. PDLIM2 inhibits T helper 17 cell development and granulomatous inflammation through degradation of STAT3. Sci. Signal., 4, ra85 (2011).
- 37) Strassmann G, Patil-Koota V, Finkelman F, Fong M, Kambayashi T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J. Exp. Med., 180, 2365–2370 (1994).
- 38) Németh K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med., 15, 42–49 (2009).
- 39) Shang L, Lye DC, Cao B. Contemporary narrative review of treatment options for COVID-19. Respirology, 26, 745–767 (2021) doi: 10.1111/resp.14106.
- 40) Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med., 11, eaaw1736 (2019).
- 41) Wrobleski ST, Moslin R, Lin S, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J. Med. Chem., 62, 8973–8995 (2019).
- 42) Moslin R, Zhang Y, Wrobleski ST, et al. Identification of N-methyl nicotinamide and N-methyl pyridazine-3-carboxamide pseudokinase domain ligands as highly selective allosteric inhibitors of tyrosine kinase 2 (TYK2). J. Med. Chem., 62, 8953–8972 (2019).
- 43) Chang Y, Xu S, Ding K. Tyrosine kinase 2 (TYK2) allosteric inhibitors to treat autoimmune diseases. J. Med. Chem., 62, 8951–8952 (2019).
- 44) Fensome A, Ambler CM, Arnold E, et al. Dual inhibition of TYK2 and JAK1 for the treatment of autoimmune diseases: discovery of ((S)-2,2-difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1] octan-8-yl)methanone (PF-06700841). J. Med. Chem., 61, 8597–8612 (2018).
- 45) Pellenz FM, Dieter C, Lemos NE, Bauer AC, Souza BM, Crispim D. Association of TYK2 polymorphisms with autoimmune diseases: A comprehensive and updated systematic review with meta-analysis. Genet. Mol. Biol., 44, e20200425 (2021).
- 46) Li Z, Gakovic M, Ragimbeau J, Eloranta ML, Rönnblom L, Michel F, Pellegrini S. Two rare disease-associated Tyk2 variants are catalytically impaired but signaling competent. J. Immunol., 190, 2335–2344 (2013).