Regular Articles
Increased TMEM16A-Mediated Ca2+-Activated Cl− Currents in Portal Vein Smooth Muscle Cells of Caveolin 1-Deficient Mice
2022 年 45 巻 11 号 p. 1692-1698

詳細
2022 年 45 巻 11 号 p. 1692-1698
Ca2+-activated Cl− (ClCa) channels regulate membrane excitability and myogenic tone in vascular smooth muscles. TMEM16A-coding proteins are mainly responsible for functional ClCa channels in vascular smooth muscles, including portal vein smooth muscles (PVSMs). Caveolae are cholesterol-rich and Ω-shaped invaginations on the plasma membrane that structurally contributes to effective signal transduction. Caveolin 1 (Cav1) accumulates in caveolae to form functional complexes among receptors, ion channels, and kinases. The present study examined the functional roles of Cav1 in the expression and activity of ClCa channels in the portal vein smooth muscle cells (PVSMCs) of wild-type (WT) and Cav1-knockout (KO) mice. Contractile experiments revealed that the amplitude of spontaneous PVSM contractions was larger in Cav1-KO mice than WT mice. Under whole-cell patch-clamp configurations, ClCa currents were markedly inhibited by 1 µM Ani9 (a selective TMEM16A ClCa channel blocker) in WT and Cav1-KO PVSMCs. However, Ani9-sensitive ClCa currents were significantly larger in Cav1-KO PVSMCs than in WT PVSMCs. Expression analyses showed that TMEM16A expression levels were higher in Cav1-KO PVSMs than in WT PVSMs. Therefore, the caveolar structure formed by Cav1 negatively regulates the expression and activity of TMEM16A-mediated ClCa channels in vascular smooth muscle cells.
Cl− channels are ubiquitously expressed to regulate Cl− homeostasis, membrane excitability, cell volume, and survival. The Cl− channel superfamily is structurally classified into four groups: voltage-dependent Cl− channels, cystic fibrosis transmembrane conductance regulator channels, Ca2+-activated Cl− (ClCa) channels, and ligand-gated Cl− channels.1,2) The conductance of ClCa channels is involved in several physiological functions, such as epithelial secretions, neuronal transmission, sensory transduction, nociceptive responses, and muscle contraction.3) ClCa channel activity is regulated by the cytosolic Ca2+ concentration ([Ca2+]cyt), the membrane potential, and calmodulin. In vascular smooth muscles, ClCa channel conductance regulates the membrane excitability, cell proliferation, and myogenic response.4)
Two proteins belonging to the TMEM16/anoctamin family, TMEM16A/ANO1 and TMEM16B/ANO2, were recently identified as ClCa channels in several tissues. TMEM16A ClCa channels are widely distributed in many types of cells, such as epithelial cells, nociceptive neurons, the interstitial cells of Cajal, and smooth muscle cells.3) TMEM16A ClCa channels expressed in vascular smooth muscles of the aorta, basilar artery, mesenteric artery, pulmonary artery, and portal vein. TMEM16B ClCa channels are localized to hippocampal neurons, olfactory neurons, retinal photoreceptors,3) and pineal glands.5)
Caveolae are Ω-shaped membrane invaginations (50 to 100 nm in diameter) that are enriched in cholesterol and sphingolipids. As the structural proteins required for the formation of caveolar membrane domains, three caveolin proteins (caveolin 1, 2, and 3) have been cloned.6,7) Caveolin 1 (Cav1) accumulates in caveolae and recruits different signaling molecules (e.g., receptors, ion channels, and kinases) during physiological and pathological responses. This Cav1-mediated compartmentalization results in efficient signal transduction following agonistic and specific stimuli. A previous study reported that large-conductance Ca2+-activated K+ (BKCa) channels were preferentially distributed in caveolae over non-caveolar regions on the membrane surface in rat aortic myocytes.8) Furthermore, voltage-dependent Ca2+ channels (VDCCs) were shown to aggregate in caveolae and functionally interact with BKCa channels in order to regulate membrane excitability in murine mesenteric myocytes.9) Functional interactions and molecular complexes in caveolae may be essential for a microdomain of Ca2+ signaling (e.g., Ca2+ sparks and Ca2+ hotspots) in vascular smooth muscle cells.10)
In contrast to BKCa channels and VDCCs, limited information is currently available on the involvement of caveolae and caveolins in the expression and activity of ClCa channels. The deletion of cholesterol was previously shown to increase TMEM16A-mediated ClCa currents in murine portal vein smooth muscle cells (PVSMCs).11) In contrast, the addition of cholesterol decreased the expression and activity of TMEM16A-mediated ClCa channels in human aortic endothelial cells.12)
In the present study, using wild-type (WT) and Cav1-knockout (KO) mice, the functional roles of Cav1 in the expression and activity of ClCa channels in PVSMCs were examined by contractile measurements, whole-cell patch-clamp recordings, quantitative real-time PCR, and Western blotting. The results obtained demonstrated that spontaneous contractions and ClCa currents were larger in Cav1-KO PVSMCs than in WT PVSMCs, and the expression of TMEM16A ClCa channels was upregulated in the PVSMCs of Cav1-KO mice.
All experiments were approved by the Ethics Committee of Nagoya City University (H30-P-1) and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Japanese Pharmacological Society.
Contractile MeasurementsThe portal vein was dissected from WT mice (C57BL/6, male/female, 8 to 16 weeks old; Japan SLC, Hamamatsu, Japan) and Cav1-KO mice (C57BL/6 background, male/female, age-matched; Jackson Laboratory, Bar Harbor, ME, U.S.A.) in aerated Krebs’ solution (112 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 25 mM NaHCO3, 1.2 mM KH2PO4, 14 mM glucose, and pH 7.4 by gassing with a mixture of 95% O2/5% CO2) and the endothelium was stripped out by water flow.13) Portal vein smooth muscles (PVSMs; length of 5 mm) were set in a tissue chamber filled with aerated Krebs’ solution at 37 °C and then stretched to 1 mN of tension. Contractile responses were measured using an isometric transducer (UM-203; Iwashiya Kishimoto Medical Instruments, Kyoto, Japan) and the data acquisition system (PowerLab 2/26; ADInstruments, Bella Vista, Australia).
Cell IsolationPVSMs were incubated in physiological saline solution (PSS; 125 mM NaCl, 5.4 mM KCl, 0.05 mM CaCl2, 15.4 mM NaHCO3, 0.33 mM Na2HPO4, 0.34 mM KH2PO4, 10 mM glucose, and 11 mM N-(2-hydroxyethyl) piperazine-N′-2-ethanesulfonic acid (HEPES)) containing 0.3% protease (type XIV; Sigma-Aldrich, St. Louis, MO, U.S.A.) at 37 °C for 5 min.13) Tissues were then incubated in PSS containing 0.6% collagenase (type IA; Sigma-Aldrich) at 37 °C for 5 min. After the incubation, single PVSMCs were obtained by mechanical dispersion in PSS.
Electrophysiological RecordingsElectrophysiological recordings were performed on single PVSMCs by a whole-cell patch-clamp technique with a CEZ-2400 amplifier (Nihon Kohden, Tokyo, Japan), analog-digital converter (Digidata 1440 A; Axon-Molecular Devices, Foster City, CA, U.S.A.), and pCLAMP software (version 10; Axon-Molecular Devices) in HEPES-buffered extracellular solution (137 mM NaCl, 5.9 mM KCl, 2.2 mM CaCl2, 1.2 mM MgCl2, 14 mM glucose, 10 mM HEPES, and pH 7.4 with NaOH) at room temperature (23 to 25 °C).14) The pipette solution had the following composition: 120 mM CsCl, 20 mM tetraethylammonium (TEA) chloride, 2.8 mM MgCl2, 2 mM ATPNa2, 4.25 mM CaCl2, 5 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N,′N′-tetraacetic acid (EGTA) (pCa 6.0), 10 mM HEPES, and pH 7.2 with CsOH.
Quantitative Real-Time PCRmRNA expression levels in PVSMs were examined using the real-time PCR system (ABI PRISM 7000; Applied Biosystems, Foster City, CA, U.S.A.).13) Specific primers were designed: Tmem16a (NM_178642, 2583–2715), Tmem16b (NM_153589, 2779–2899), Tmem16c (NM_001128103, 2716–2827), Tmem16d (NM_001277188, 3121–3233), Tmem16e (NM_177694, 2492–2592), Tmem16f (NM_001253813, 2592–2697), Tmem16g (NM_207031, 2338–2442), Tmem16h (NM_001164679, 2429–2536), Tmem16j (NM_178381, 2096–2199), Tmem16k (NM_133979, 1735–1836), and Gapdh (glyceraldehyde-3-phosphate dehydrogenase; NM_001289726, 776–879). Values for each unknown sample relative to the standard curve for specific primers were calculated and yielded transcriptional quantitation of gene products relative to the endogenous standard (Gapdh).
Western BlottingThe protein fraction was extracted from PVSMs using sample buffer (62.5 mM Tris–HCl, 2% sodium dodecyl sulfate (SDS), and 10% glycerol). Extracted protein (40 µg/lane) was subjected to 7.5% SDS-polyacrylamide gel electrophoresis (PAGE) (BioRad, Hercules, CA, U.S.A.) and transferred to a nitrocellulose membrane (Millipore, Bedford, MA, U.S.A.).5) The resulting blots were treated with 5% fat-free milk (Megmilk Snow Brand, Tokyo, Japan) in TBS-T solution (Tris-buffered saline containing 0.1% Tween 20) at room temperature for 1 h and incubated with an anti-TMEM16A antibody (1 : 100; ab53212, Abcam, Cambridge, U.K.) at 4 °C for 12 h. After the membrane was washed with TBS-T solution for 30 min, it was treated with an anti-rabbit horseradish peroxidase-conjugated immunoglobulin G (IgG) antibody (1 : 2000; #7074, Cell Signaling Technology, Danvers, MA, U.S.A.) at room temperature for 1 h and rinsed with TBS-T solution for 30 min. Protein expression was detected using the ECL Prime detection reagent (Amersham Biosciences, Piscataway, NJ, U.S.A.) and the Imager 600 system (Amersham Biosciences). Protein expression was normalized using anti-β-actin antibody (1 : 5000; A1978, Sigma-Aldrich) and anti-mouse horseradish peroxidase-conjugated IgG antibody (1 : 2000; #7076, Cell Signaling Technology).
DrugsPharmacological reagents were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), except for Ani9 (2-(4-chloro-2-methylphenoxy)-N-[(2-methoxyphenyl)methylideneamino]-acetamide; Sigma-Aldrich), EGTA, HEPES (Dojindo Laboratories, Kumamoto, Japan), and TEA chloride (Tokyo Chemical Industry, Tokyo, Japan). Ani9 was dissolved in dimethyl sulfoxide at a concentration of 10 mM as a stock solution.
Statistical AnalysisPooled data are shown as the mean ± standard error (S.E.). The significance of differences between two groups was assessed by the Student’s t-test using BellCurve for Excel software (version 3.22; Social Survey Research Information, Tokyo, Japan).
PVSMs exhibit spontaneous and periodic contractions without any stimulations and spontaneous contractions are sensitive to ClCa blockers.13) To demonstrate the direct involvement of Cav1 in spontaneous PVSM contractions, contractile responses were recorded in the PVSMs of WT and Cav1-KO mice. In WT PVSMs, the mean amplitude of spontaneous contractions was 0.163 ± 0.030 mN (n = 5; Figs. 1A, B) and the mean frequency was 33.6 ± 6.4 min−1 (n = 5; Figs. 1A, C). We previously reported that spontaneous contractions were reduced by 30 µM T16Ainh-A01 (a specific TMEM16A ClCa channel blocker).13) In Cav1-KO PVSMs, their amplitude was increased (0.345 ± 0.071 mN, n = 4, p = 0.038; Figs. 1A, B), whereas their frequency was not altered (25.9 ± 3.1 min−1, n = 4, p = 0.357; Figs. 1A, C). These results suggest that the spontaneous contractions of PVSMs, which were mainly mediated by TMEM16A ClCa channels, were negatively regulated by Cav1.

The functional roles of Cav1 in spontaneous PVSM contractions were examined using WT and Cav1-KO mice. A. Representative recordings of spontaneous contractions without any stimulation in WT and Cav1-KO PVSMs. B. The mean amplitude of spontaneous contractions in WT and Cav1-KO PVSMs (n = 4 to 5 mice). C. The mean frequency of spontaneous contractions in WT and Cav1-KO PVSMs (n = 4 to 5 mice). * p < 0.05 vs. WT.
Under whole-cell voltage-clamp conditions, ClCa currents were measured in the PVSMCs of WT and Cav1-KO mice using K+-deficient pipette solution containing 120 mM Cs+ and 20 mM TEA. The Ca2+ concentration in the pipette solution was fixed to pCa 6.0 (1 µM) for the activation of ClCa currents. Single PVSMCs were stimulated from the holding potential of −60 mV to the test potentials (from −100 to +80 mV) by +20-mV increments for 1 s and were then repolarized to −80 mV for 500 ms every 15 s. The mean cell capacitance of WT PVSMCs was 36.8 ± 3.1 pF (n = 12). A depolarizing pulse caused time-dependent outward currents in WT PVSMCs (10.3 ± 0.8 pA/pF at +80 mV, n = 12; Figs. 2A, B). At repolarization at −80 mV following +80 mV depolarization, inward tail currents were observed (−11.2 ± 1.1 pA/pF, n = 12). We previously reported that these currents were largely blocked by the application of 100 µM niflumic acid (a conventional ClCa channel blocker) and 10 to 30 µM T16Ainh-A01.15) In the present study, the effects of Ani9, which was recently shown to function as a potent and selective blocker of TMEM16A ClCa channels,16) on ClCa currents were examined in PVSMCs. The outward peak and inward tail currents were decreased by the application of 1 µM Ani9 (4.4 ± 0.6 pA/pF at +80 mV, n = 12, p < 0.001 vs. control and −2.9 ± 0.4 pA/pF at −80 mV after +80 mV depolarization, n = 12, p < 0.001, respectively). The inhibitory effect was mostly recovered by the removal of Ani9 (n = 12).
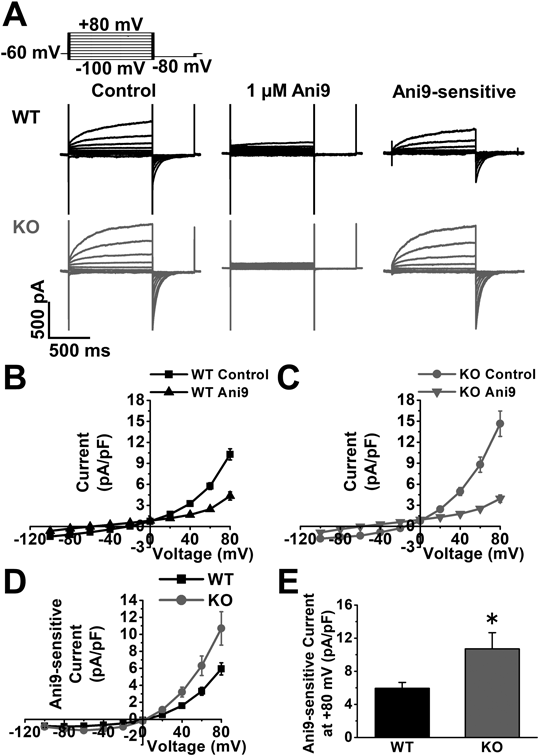
ClCa currents were recorded in the PVSMCs of WT and Cav1-KO mice. Single PVSMCs were stimulated from a holding potential of −60 mV to selected test potentials (from −100 to +80 mV) by +20-mV increments for 1 s and were then repolarized to −80 mV for 500 ms every 15 s. A. Representative traces of ClCa currents in the absence and presence of 1 µM Ani9, and Ani9-sensitive currents in WT and Cav1-KO PVSMCs. B. The current-voltage relationship of ClCa currents with and without Ani9 in the PVSMCs of WT mice (n = 12 cells). C. The current-voltage relationship of ClCa currents with and without Ani9 in the PVSMCs of Cav1-KO mice (n = 13 cells). D. The current-voltage relationship of Ani9-sensitive ClCa currents in WT and Cav1-KO PVSMCs (n = 12 to 13 cells). E. The peak amplitude of Ani9-sensitive ClCa currents at +80 mV in WT and Cav1-KO PVSMCs (n = 12 to 13 cells). * p < 0.05 vs. WT.
The activity of ClCa currents was then measured in the PVSMCs of Cav1-KO mice. Cell capacitance (30.6 ± 2.0 pF, n = 13, p = 0.097) was identical to that in the PVSMCs of WT mice. However, membrane depolarization elicited larger outward peak currents (14.6 ± 1.8 pA/pF at +80 mV, n = 13, p = 0.042 vs. WT) and inward tail currents (−17.3 ± 2.4 pA/pF at −80 mV after +80 mV depolarization, n = 13, p = 0.032) in the PVSMCs of Cav1-KO mice (Figs. 2A, C). Outward peak and inward tail currents were reduced by 1 µM Ani9 (3.9 ± 0.5 pA/pF at +80 mV, n = 13, p < 0.001 vs. control and −3.1 ± 0.7 pA/pF at −80 mV after +80 mV depolarization, n = 13, p < 0.001, respectively). Ani9-sensitive ClCa currents were larger in Cav1-KO PVSMCs (10.7 ± 2.0 pA/pF at +80 mV, n = 13, p = 0.038) than in WT PVSMCs (5.9 ± 0.7 pA/pF, n = 12; Figs. 2A, D, E). The current-voltage relationship clearly showed that reversal potentials were approximately 0 mV in WT and Cav1-KO PVSMCs (n = 12 to 13; Fig. 2D), which was the theoretical equilibrium potential of Cl− under these experimental conditions. These results indicate that the activity of ClCa currents was negatively regulated by Cav1 in PVSMCs.
Electrophysiological properties of whole-cell ClCa currents were compared between WT and Cav1-KO PVSMCs. No significant differences were observed in the inhibitory effects of Ani9 at +40, +60, or +80 mV depolarization between WT and Cav1-KO PVSMCs (58 ± 5% decrease at +80 mV, n = 12 and 69 ± 5% decrease, n = 13, respectively, p = 0.126; Fig. 3A). The time constant for the activation (τact) of ClCa currents in Cav1-KO PVSMCs (280 ± 16 ms at +80 mV, n = 13, p = 0.441) was similar to that in WT PVSMCs (294 ± 7 ms, n = 12; Fig. 3B). The time constant for the deactivation (τdeact) of ClCa currents was slightly slower in Cav1-KO PVSMCs (39 ± 5 ms at −80 mV after +80 mV depolarization, n = 13, p = 0.045) than in WT PVSMCs (22 ± 5 ms, n = 12; Fig. 3C). These results suggest that ClCa currents increased in Cav1-KO PVSMCs without changes in Ani9 sensitivity or activation kinetics.

The electrophysiological and pharmacological characteristics of ClCa currents were compared between WT and Cav1-KO PVSMCs. ClCa current data obtained in Fig. 2 were reanalyzed. A. Inhibitory effects of 1 µM Ani9 at +40, +60, and +80 mV depolarization in WT and Cav1-KO PVSMCs (n = 12 to 13 cells). B. τact of ClCa currents at +80 mV in WT and Cav1-KO PVSMCs (n = 12 to 13 cells). C. τdeact of ClCa currents at −80 mV following +80 mV depolarization in WT and Cav1-KO PVSMCs (n = 12 to 13 cells). * p < 0.05 vs. WT.
The expression profile of the TMEM16 family in the PVSMs of WT and Cav1-KO mice was examined by quantitative real-time PCR and Western blot analyses. Tmem16a mRNA was highly expressed in WT PVSMs (0.081 ± 0.010, n = 6; Fig. 4A), which is consistent with previous findings.13,15) Tmem16f and Tmem16k mRNAs were also detected in WT PVSMs (0.057 ± 0.015 and 0.070 ± 0.018, respectively, n = 6). In Cav1-KO PVSMs, the mRNA expression of Tmem16a was upregulated (0.182 ± 0.027, n = 6, p = 0.006). Tmem16f mRNA levels were slightly increased in Cav1-KO PVSMs (0.098 ± 0.005, n = 6, p = 0.031), whereas Tmem16k mRNA levels were similar to those in WT PVSMs (0.082 ± 0.004, n = 6, p = 0.516). No significant differences were observed in other Tmem16 genes between WT and Cav1-KO PVSMs (n = 6, p > 0.050). TMEM16A protein expression levels were also higher in Cav1-KO PVSMs (1.24 ± 0.07-fold, n = 6, p = 0.047) than in WT PVSMs (1.00 ± 0.08, n = 6; Figs. 4B, C). Collectively, these results demonstrate that the expression of TMEM16A ClCa channels at the mRNA and protein levels were higher in Cav1-KO PVSMs than in WT PVSMs, resulting in increased TMEM16A-mediated ClCa currents and spontaneous contractions in the PVSMCs of Cav1-KO mice.
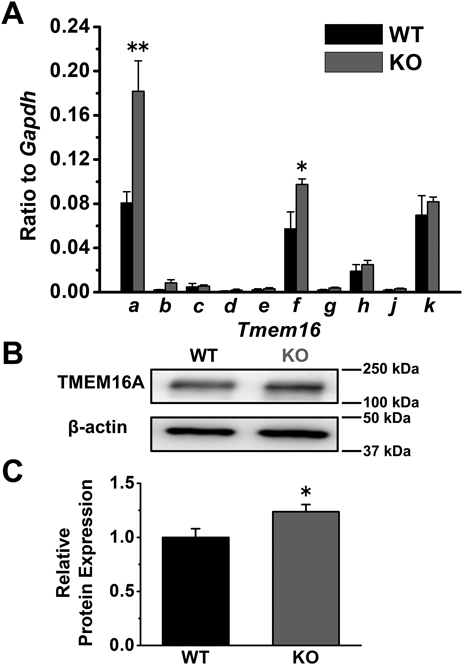
The expression levels of the TMEM16 family in the PVSMs of WT and Cav1-KO mice were qualified by quantitative real-time PCR and Western blot analyses. A. Tmem16 mRNA expression levels in the PVSMs of WT and Cav1-KO mice (n = 6 mice). Expression values were normalized to Gapdh. B. Representative blots showing TMEM16A protein expression in the PVSMs of WT and Cav1-KO mice. C. TMEM16A protein expression in WT and Cav1-KO PVSMs (n = 6 mice). Protein levels were normalized to the expression of β-actin and WT PVSMs. * p < 0.05, ** p < 0.01 vs. WT.
We herein demonstrated that spontaneous contractions and ClCa currents were increased by the upregulated expression of TMEM16A ClCa channels in the PVSMCs of Cav1-KO mice. To the best of our knowledge, this is the first study to report the upregulated expression of TMEM16A ClCa channels by a Cav1 deficiency in vascular smooth muscle cells.
Caveolae are a platform for the assembly of different functional proteins for efficient signal transduction. These signaling molecules (e.g., receptors, ion channels, and kinases) accumulate in caveolae and occasionally interact with each other. We previously reported that VDCCs, BKCa channels, and Ca2+/calmodulin-dependent kinases interacted with Cav1 and localized in caveolae, leading to efficient signal transduction as a Ca2+ microdomain in rat aortic and murine mesenteric arterial myocytes.8,9,17) However, previous findings also suggest the involvement of caveolae and caveolins in the expression and activity of ClCa channels. In the present study, the amplitude of spontaneous PVSM contractions, which are mediated by TMEM16A-mediated ClCa channels,13) was increased in Cav1-KO mice (Fig. 1). This result indicates that the activity of TMEM16A-mediated ClCa channels was enhanced by deficiencies of Cav1 and caveolae in vascular smooth muscle cells.
Similar to spontaneous contractions, larger ClCa currents were observed in the PVSMCs of Cav1-KO mice. Importantly, ClCa currents sensitive to Ani9 (a selective blocker of TMEM16A ClCa channels)16) were larger in Cav1-KO PVSMCs than in WT PVSMCs. This result (Fig. 2) was consistent with those obtained in contractile experiments (Fig. 1).Vascular tone is mainly influenced by the balance between BKCa and ClCa channel conductance. It is finely regulated by Ca2+ sparks, which are localized and spontaneous Ca2+ release from ryanodine receptors on the junctional sarcoplasmic reticulum, under physiological conditions.10) In vascular smooth muscle cells, Ca2+ sparks activate plasmalemmal BKCa channels and elicit spontaneous transient outward currents (STOCs), resulting in membrane hyperpolarization. This reduces VDCC activity, decreases [Ca2+]cyt, and relaxes vascular smooth muscles. In contrast, the activation of plasmalemmal ClCa channels by Ca2+ sparks causes spontaneous transient inward currents (STICs). STICs induce membrane depolarization, subsequent increase in [Ca2+]cyt, and, ultimately, vascular smooth muscle contractions. STOCs and STICs have been simultaneously observed in several myocytes of the trachea18) and corpus cavernosum.19) Ca2+ sensitivity between BKCa and ClCa channels is in a similar concentration range (EC50 = 1 to 10 µM).3,20) In addition, BKCa channels were found to accumulate in caveolae near junctional sarcoplasmic reticulum, at which Ca2+ sparks frequently occur.10) ClCa channels have been suggested to aggregate in close proximity to Ca2+ spark sites.21) Therefore, vascular tone is maintained by the opposite activities of BKCa and ClCa channels.
Although ClCa currents were larger in the PVSMCs of Cav1-KO mice (Fig. 2), no significant differences were observed in morphological (cell capacitance), electrophysiological (τact), or pharmacological (Ani9 sensitivity) parameters between WT and Cav1-KO PVSMCs (Fig. 3). The present results strongly suggest that the increased activity of ClCa currents in Cav1-KO PVSMCs was elicited by the upregulated expression of TMEM16A ClCa channels rather than by the facilitated activation of the TMEM16A ClCa channel itself. This hypothesis was supported by the results of the expression analyses revealing that the expression of TMEM16A ClCa channels at the mRNA and protein levels was upregulated in the PVSMs of Cav1-KO mice (Fig. 4). The expression of TMEM16A ClCa channels was previously shown to be upregulated in essential hypertension22) and pulmonary arterial hypertension,23) but downregulated in renovascular hypertension24) and cirrhotic portal hypertension.13) Interestingly, Cav1-KO mice exhibit pulmonary arterial hypertension.6,25) In the present study, the mechanisms by which TMEM16A expression was upregulated in the PVSMCs of Cav1-KO mice were not elucidated. More recently, we demonstrated that an exposure to angiotensin II, which increases in cirrhotic portal hypertension, downregulated the expression of TMEM16A ClCa channels in PVSMCs.13) However, other findings showed that angiotensin II upregulated its expression in rat mesenteric myocytes, contributing to the pathological mechanism of essential hypertension.22) Further studies are needed to elucidate the mechanism underlying the upregulation of TMEM16A ClCa channels in vascular smooth muscle cells of Cav1-KO mice.
Expression analyses of the TMEM16 family showed that the Tmem16a/Ano1, Tmem16f/Ano6, and Tmem16k/Ano10 genes were highly expressed in PVSMs (Fig. 4), which is consistent with previous findings.13,15) In addition to Tmem16a (225%), Tmem16f transcripts increased in the PVSMs of Cav1-KO mice (170%). TMEM16F is recognized as a Ca2+-dependent phospholipid scramblase, but may form a Cl− channel that requires high [Ca2+]cyt for its activation (EC50 of >10 µM).3) Tmem16k (118%) is not considered to be an ion channel.3) Therefore, we examined the upregulation of TMEM16A ClCa channels in the PVSMs of Cav1-KO mice. The cytosolic Cl− concentration was previously reported to be 30 to 50 mM in vascular smooth muscle cells4); therefore, the reversal potential of Cl− was calculated to be −40 to −25 mV. Since the resting membrane potential is −60 to −40 mV in vascular smooth muscle cells,1) the activation of Cl− channels (due to the upregulation of Cl− channels) causes membrane depolarization and facilitates Ca2+ influx through VDCCs. This pathway may contribute to the enhancement of spontaneous PVSM contractions in Cav1-KO mice.
Caveolins are scaffolding proteins that assemble different signaling molecules as functional complexes in caveolae, which are lipid raft domains on the membrane surface. In caveolae, several signal molecules and downstream cascades are modulated by an interaction with caveolin proteins to facilitate efficient signal transduction. Cav1 proteins are abundantly expressed in smooth muscle cells, endothelial cells, cardiac fibroblasts, and macrophages.6) Therefore, structural disruptions caused by the ablation of caveolins result in cardiovascular diseases, including diabetes, pulmonary hypertension, and atherosclerosis.6) Besides, Cav1 mutations have been associated with congenital generalized lipodystrophy type 3 (also known as Berardinelli–Seip syndrome), pulmonary arterial hypertension, and breast cancer.7) Cav1 has been shown to modulate the activity of volume-regulated Cl− channels (leucine-rich repeat-containing 8) in reconstituted human embryonic kidney 293 cells26) and voltage-dependent Cl− channels (ClC-2) in human intestinal Caco-2 cells.27) In addition, we herein showed that a Cav1 deficiency induced the upregulation of TMEM16A ClCa channels in vascular smooth muscle cells. This effect of Cav1 on the expression and activity of TMEM16A ClCa channels may be informative for elucidating the pathophysiological significance of vascular TMEM16A ClCa channels.
In conclusion, TMEM16A-mediated ClCa channels were negatively regulated by Cav1 and/or caveolar structure in vascular smooth muscle cells. Since TMEM16A ClCa channels contribute to the regulation of membrane excitability, contraction, and proliferation/apoptosis in vascular smooth muscle cells, a functional analysis of TMEM16A ClCa channels is important for elucidating their physiological and pathological significance.
The present study was supported by Grants-in-Aid for Scientific Research (B) (22H02773 to Y. Suzuki; 22H02787 to H. Yamamura) from the Japan Society for the Promotion of Science. N. Kawata has an ENPHAS scholarship from Nagoya City University. We are grateful for the assistance of the Research Equipment Sharing Center at Nagoya City University.
The authors declare no conflict of interest.