Abstract
The long-term administration of tamoxifen to estrogen receptor α (ERα)-positive breast cancer patients is an established treatment that reduces mortality and recurrence. However, resistance to tamoxifen and an increased risk of endometrial cancer may occur; therefore, the mechanisms by which tamoxifen causes these adverse effects warrant further study. Tamoxifen has been shown to activate mitogen-activated protein kinase (MAPK) in an ERα-independent manner; therefore, we investigated its effects on the MAPK-mediated non-canonical activation of EphA2, a critical event regulating cell migration. Tamoxifen at slightly higher concentrations induced the rapid phosphorylation of EphA2 at Ser-897 via the MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK)–ERK–ribosomal S6 kinases (RSK) pathway in HeLa cells. In addition, tamoxifen significantly enhanced the migration ability of ERα-negative MDA-MB-231 breast cancer cells in RSK- and EphA2-dependent manners. Phosphorylated EphA2 was internalized and re-localized to the plasma membrane, including lamellipodia, in an RSK-dependent manner. Collectively, the present results provide novel insights into the tumor-promoting activity of tamoxifen.
INTRODUCTION
Tamoxifen is an estrogen receptor α (ERα) modulator that has been used as a first-line treatment for ERα-positive breast cancer patients. Although most patients benefit from tumor reductions and the suppression of metastasis, some patients are resistant to tamoxifen.1,2) Furthermore, tamoxifen has been shown to increase the risk of endometrial tumor development,3) distant metastasis, and other adverse effects.4–6)
ERα is a transcription factor that is activated by estrogen binding. Tamoxifen competes with the binding of estrogen to ERα, which suppresses the transcriptional activity of ERα.7) In addition to its traditional activity in the nucleus, recent studies reported that tamoxifen, as well as estrogen, at slightly elevated concentrations rapidly activated the mitogen-activated protein kinase (MAPK) signaling pathway in the cytoplasm.8–10) In addition, tamoxifen and estrogen induced cytoskeletal remodeling and migration in endometrial cancer cells.11) Therefore, tamoxifen may exhibit tumor-promoting activity despite its expected effectiveness for tumor suppression.12,13) However, the exact mechanisms by which tamoxifen induces tumorigenesis and metastasis have not yet been elucidated.
EphA2 belongs to the Eph receptor subfamily and is frequently overexpressed in several cancer types.14,15) The ligands of EphA2, including Ephrin-A1, induce the canonical tyrosine (Tyr) kinase activation of EphA2, which is involved in its tumor suppressor functions.16,17) On the other hand, epidermal growth factor (EGF) and inflammatory cytokines, such as tumor necrosis factor (TNF)-α, induce the non-canonical activation of EphA2 via phosphorylation at C-terminal Ser-897.18–20) We originally reported that the phosphorylation of Ser-897 was directly catalyzed by ribosomal S6 kinases (RSKs), downstream molecules of extracellular signal-regulated kinase (ERK) MAPK.20) Protein kinase B (Akt) and protein kinase A (PKA) are also known as kinases for Ser-897.21) The atypical activation of EphA2 promotes cell motility, which correlates with a poor prognosis in lung cancer patients.20) However, the effects of tamoxifen on the activation of EphA2 remain unclear.
In the present study, we confirmed the activation of ERK by tamoxifen, and revealed that the RSK-EphA2 axis is crucial for promoting cell migration by tamoxifen in ERα-negative breast cancer cells.
MATERIALS AND METHODS
Antibodies and ReagentsAntibodies against EphA2 (D4A2; #6997), phospho-EphA2 (Ser-897; #6347), phospho-RSK (Ser-380; #11989), and phospho-ERK (Thr-202/Tyr-204; #9101) were obtained from Cell Signaling Technology (Danvers, MA, U.S.A.). RSK1 (C-21; sc-231). RSK2 (C-19; sc-1430), ERK (E-6; sc-271270), β-actin (I-19; sc-1616), and α-tubulin (B-7; sc-5286) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Human recombinant EGF and TNF-α were purchased from R&D Systems (Minneapolis, MN, U.S.A.). 4-Hydroxy tamoxifen and trametinib were obtained from Cayman Chemical (Ann Arbor, MI, U.S.A.). BI-D1870 was obtained from BioVision (Milpitas, CA, U.S.A.). All chemical inhibitors were dissolved in dimethyl sulfoxide (DMSO).
Cell CultureCell cultures were performed as previously described.20) Briefly, HeLa, HEK293, and MDA-MB-231 cells were obtained from the American Type Culture Collection (ATCC, Rockville, TX, U.S.A.) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (high-glucose condition; Life Technologies Corporation, Carlsbad, CA, U.S.A.) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 U/mL streptomycin (Meiji Seika Pharma, Tokyo, Japan) at 37 °C in 5% CO2. Regarding the tamoxifen treatment, cells were maintained in phenol red and serum-free medium for 24 h before experimentation.
ImmunoblottingWhole-cell lysates were prepared as previously described.20,22,23) Briefly, cells were lysed in lysis buffer containing 1 mM dithiothreitol (DTT), 1 mM sodium orthovanadate, 20 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 µg/mL leupeptin, and 10 µg/mL aprotinin. Each lysate was mixed with the same volume of sample buffer for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (100 mM Tris–HCl (pH 6.8), 70 mM DTT, 2.0% SDS, 10% glycerol, and 0.10% bromophenol blue) and heated at 95 °C for 5 min. Lysates were resolved by SDS-PAGE and transferred to an Immobilon-P membrane (Millipore, Billerica, MA, U.S.A.). The membrane was incubated with BlockAce overnight (KAC Co., Ltd., Kyoto, Japan), treated with primary antibodies, and then incubated with horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) (P0448; diluted 1 : 2000), anti-mouse IgG (P0260; diluted 1 : 2000), or anti-goat IgG (P0449; diluted 1 : 2000) (DAKO, Glostrup, Denmark). Antibodies were visualized with the ECL system (GE Healthcare Bioscience, Piscataway, NJ, U.S.A.). Primary antibody reactions were performed in Can Get Signal solution (TOYOBO, Tokyo, Japan) and second antibody reactions in phosphate buffered saline (PBS) containing 0.1% Tween 20 (FUJIMILM Wako Pure Chemical Corporation, Osaka, Japan).
RNA Interference (RNAi)Small interfering RNAs (siRNAs) were synthesized at Thermo Fisher Scientific, Inc., Waltham, MA, U.S.A. (Stealth RNAi). Target sequences were as follows: RSK1 (5′-CCAUGCUGGCAGGAUAUACUCCAUU-3′), RSK2 (5′-GGGAGGAGAUUUGUUUACACGCUUA-3′), and EphA2 (5′-GCAAGGAAGUGGUACUGCUGGACUU-3′). siRNA Negative Control Lo GC Duplex #2 was used. HeLa cells were transfected with siRNAs at a final concentration of 100 nM using Lipofectamine Reagent (Thermo Fisher Scientific, Inc.). Cells were incubated for 72 h before experiments.
ImmunofluorescenceImmunofluorescence was performed as previously described.22) Briefly, cells were seeded on glass cover slips (Thermo Fisher Scientific, Inc.) 24 h before treatment, and then treated with inhibitors and ligands, washed with cold PBS, and fixed in paraformaldehyde at room temperature for 15 min. After fixation, cells were treated with PBS containing 0.5% Triton X-100. Cells were treated with primary antibodies for 40 min and then incubated with Alexa Fluor 488-conjugated secondary antibodies (Thermo Fisher Scientific, Inc.) for 30 min. Primary and secondary antibodies were diluted in PBS containing 0.5% bovine serum albumin (BSA). The coverslips were flipped onto slides with 4′-6-diamidino-2-phenylindole (DAPI)-containing SlowFade Gold Antifade Reagent (Thermo Fisher Scientific, Inc.). Fluorescence microscopy was performed using a confocal microscope (LSM 700, Zeiss, Oberkochen, Germany).
Time-Lapse MicroscopyCells were treated with inhibitors and ligands and then continuously photographed every 5 min for 2 h in a CO2 incubator (37 °C, 5% CO2) using a time-lapse imaging system (Carl Zeiss Cell Observer). The images were acquired using an imaging system. The resulting images were processed by Manual Tracking Plugin of Image J Fiji to track the number of cells. Data on Scatter Maps and migration distances were acquired using the Chemotaxis and Migration Tool (ibidi GmbH). The significance of differences was calculated using JMP software (version 11.2.1, SAS Institute Inc., Cary, NC, U.S.A.). Differences were analyzed using a one-way ANOVA with Tukey’s post hoc test. p < 0.01 was considered to be significant.
RESULTS
Tamoxifen Induces the RSK-Mediated Phosphorylation of EphA2To investigate the effects of tamoxifen on the non-canonical activation of EphA2, HeLa cells were stimulated with tamoxifen as well as EGF and TNF-α, agents that activate the non-canonical EphA2 pathway. The treatment with tamoxifen up to 10 µM did not affect cell proliferation (data not shown), but strongly induced the phosphorylation of EphA2 at Ser-897, a target site for non-canonical activation, as well as that of ERK and RSK (Fig. 1A). The level of activation was similar to that with the TNF-α stimulation. Cells were then stimulated with different concentrations of tamoxifen to identify the effective concentration (Fig. 1B). EphA2 was activated by tamoxifen in a concentration-dependent manner and peaked at 10 µM. Therefore, we used this concentration in subsequent experiments. The time-course treatment with tamoxifen revealed that, similar to the activation kinetics of upstream ERK and RSK, EphA2 Ser-897 was rapidly phosphorylated within 5 min (Fig. 1C). The activation of ERK, RSK, and EphA2 was slightly down-regulated at 30 min and then re-activated at 60 min. Similar results were obtained in ERα-negative HEK293 cells (Fig. 1D), indicating bimodal activation by tamoxifen.
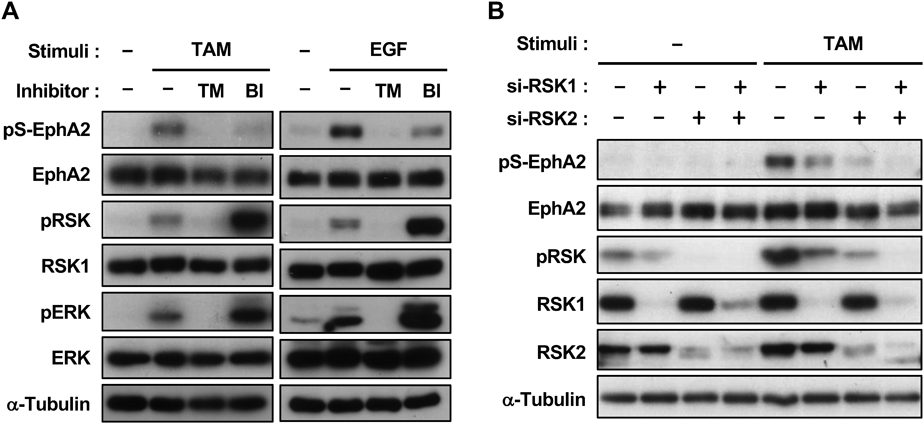
We previously demonstrated that the phosphorylation of EphA2 was mediated through the MAPK/ERK kinase–ERK–RSK pathway. Figure 2A shows that the MEK inhibitor trametinib and RSK inhibitor BI-D1870 both significantly inhibited the EGF- and tamoxifen-induced phosphorylation of EphA2, with BI-D1870 increasing the phosphorylation levels of ERK and RSK by canceling the RSK-mediated negative feedback regulation of ERK activation. Furthermore, RNAi demonstrated that the phosphorylation of EphA2 was partially suppressed by the knockdown of RSK1 or RSK2 alone, major RSK subtypes in HeLa cells, whereas double knockdown resulted in complete inhibition (Fig. 2B). Collectively, these results demonstrated that tamoxifen activated the MAPK signaling pathway, which led to the non-canonical activation of EphA2.
Tamoxifen Enhances Cell Migration in ERα-Negative Breast Cancer CellsWe used MDA-MB-231 cells to examine the effects of tamoxifen on migration properties. We and others previously reported that EphA2 promoted the migration of ERα-negative MDA-MB-231 cells, in which EphA2 is overexpressed and constitutively activated by RSK. In the present study, a time-lapse imaging system that enables individual cells to be traced was used to investigate the effects of tamoxifen on migration. Although MDA-MB-231 cells are highly metastatic, the treatment with tamoxifen further enhanced their ability to migrate. Furthermore, basal and increased cell migration abilities were suppressed by the RSK inhibitor (Fig. 3A). The siRNA knockdown of EphA2 also significantly suppressed tamoxifen-induced migration (Fig. 3B), suggesting that tamoxifen-induced cell migration is dependent on the non-canonical activation of EphA2, but not the expression of ERα.
Effects of Tamoxifen on the Intracellular Localization of EphA2Based on the results obtained in the migration assay, we investigated whether tamoxifen activated the RSK-EphA2 axis in MDA-MB-231 cells. The results obtained revealed that tamoxifen did not further activate the non-canonical EphA2 pathway, possibly because this pathway was constitutively and strongly activated (Fig. 4A). We have previously demonstrated that EphA2 is expressed on the plasma membrane, especially in the lamellipodia, a membrane structure found at the leading edge of motile cells.20) Therefore, we next examined the effect of tamoxifen on the intracellular localization of activated EphA2 by immunofluorescence. Tamoxifen induced the rapid internalization of EphA2, leading to its accumulation around the nucleus at 30–60 min, and it was then gradually recycled back to the plasma membrane, including the edge of the cell (Fig. 4B). In addition, when cells were pretreated with BI-D1870, perinuclear accumulation was not observed at 60 min, indicating that endocytosis was triggered by RSK (Fig. 4C). We also investigated the intracellular localization of Ser-897-phosphorylated EphA2 (pS-EphA2), with the specific staining of pS-EphA2 being confirmed by BI-D1870. Figure 4C shows that, similar to total EphA2 staining, tamoxifen induced the internalization of pS-EphA2 from the membrane to the perinuclear region.
DISCUSSION
Tamoxifen has a long history of use in chemotherapy for ERα-positive patients. When tamoxifen binds to the ligand-binding pocket, it exerts an antitumor effect because it is unable to attract transcriptional coactivators.7,24–26) Tamoxifen was initially considered to be an ERα antagonist; however, it was found to function as a partial ERα agonist in the uterus and bone, and, thus, is an estrogen receptor modulator (SERM).27) It also appears to exert biological effects on cells regardless of the presence of ERα.28) In the present study, we confirmed previous findings on tamoxifen-induced MAPK activation and identified the RSK-EphA2 pathway as a downstream functional component in HeLa cells. In addition, we demonstrated that the inhibition of the RSK-EphA2 pathway by an RSK inhibitor or the knockdown of EphA2 suppressed the migration of MDA-MB-231 cells (Fig. 3). Since RSKs are involved in cell migration, they have been suggested to promote migration in an EphA2-independent manner.29) However, the present results showed that RSK-EphA2 is the main signaling pathway for tamoxifen-induced cancer cell migration.
The concentration of tamoxifen used in the present study was slightly higher than that used as an estrogen antagonist. For example, a previous study reported that a stimulation with tamoxifen at 6 µM promoted the migration ability of MDA-MB-231 cells.30) Similarly, 10 µM tamoxifen induced the activation of ERK and increased migration in endometrial cancer cells.11) Therefore, the use of higher concentrations of tamoxifen is required to obtain a more detailed understanding of its wide range of biological activities.
In the present study, tamoxifen induced a transient internalization-recycling circuit of EphA2. EphA2 is a receptor tyrosine kinase that suppresses cell migration by the ligand Ephrin-A1 and has the contradictory function of promoting migration by RSK-mediated serine phosphorylation.16–20) Ligand binding suppresses Akt and MAPK, and the ligand-receptor complex is then translocated into the cell for lysosomal degradation.31,32) In contrast, the endocytosis and subsequent Rab14-dependent recycling of EphA2 are involved in cell–cell repulsion, which enables cancer cells to move away from each other and metastasize in vivo.33) Furthermore, the cleavage of the extracellular domain of EphA2 by membrane-anchored membrane type-1 matrix metalloproteinase has been shown to trigger the intracellular trafficking of EphA2 and promote the cell–cell repulsion, junctional disassembly, and dissemination of motile single cells.34) These observations suggest that the delivery of EphA2 to the leading edge via intracellular trafficking plays a key role in tamoxifen-induced cell migration. To further investigate this hypothesis, we tried to examine the effect of the knockdown of the clathrin heavy chain (CHC) in MDA-MB-231 cells. In our preliminary experiments, the knockdown resulted in the re-localization of EphA2 in the perinuclear region; however, migratory ability was not promoted (data not shown). Therefore, the major challenge is to elucidate how tamoxifen triggers EphA2 internalization-recycling circuit of EphA2. Since we demonstrated in Fig. 3B that RSK activity is required for triggering the internalization, it is possible that RSK phosphorylates EphA2 at other than Ser-897, or regulates other endocytosis-related molecules. Collectively, further studies on the sorting mechanism of EphA2 are needed to elucidate the impact of tamoxifen on cell migration.
The truncated form of ERα is considered to be the key for overcoming this issue. The expression of ERα is regulated by multiple promoters, and intermediate exons are inserted and skipped in an isoform-specific manner, resulting in numerous splice variants.35–39) ERα46 and ERα36 variants are expressed in ER-negative cancer cells, including MDA-MB-231 cells.40,41) Furthermore, these variants are re-localized near the cell surface and, thus, are considered to be involved in MAPK activation and migration by estrogen and tamoxifen, in contrast to the conventional function of full-length ERα in the nucleus.8,42) Alternatively, the G protein-coupled estrogen receptor (GPER) with seven transmembrane domains, known as GPR30, may be involved in the activation of the RSK-EphA2 pathway induced by tamoxifen.43,44)
In summary, the present study demonstrated that tamoxifen induces the atypical activation of EphA2. This result will contribute to the mechanisms underlying the cancer-promoting effects of tamoxifen being elucidated in more detail, which will increase the safety of cancer treatment with tamoxifen.
Acknowledgments
This work was supported in part by JSPS KAKENHI Grant Nos. 19H03368 and 19K23795, Moonshot R&D Grant No. JPMJMS2021, THE HOKURIKU BANK Grant-in-Aid for Young Scientists, and research Grants from Takeda Science Foundation and MSD Life Science Foundation.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol., 17, 1474–1481 (1999).
- 2) Sparano JA, Gray RJ, Ravdin PM, et al. Clinical and genomic risk to guide the use of adjuvant therapy for breast cancer. N. Engl. J. Med., 380, 2395–2405 (2019).
- 3) Chlebowski RT, Schottinger JE, Shi JX, Chung J, Haque R. Aromatase inhibitors, tamoxifen, and endometrial cancer in breast cancer survivors. Cancer, 121, 2147–2155 (2015).
- 4) Davies C, Godwin J, Gray R, Clarke M, Darby S, McGale P, Wang YC, Dowsett M, Peto R, Pan HC, Cutter D, Taylor C, Ingle J. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet, 378, 771–784 (2011).
- 5) Wei CW, Cao Y, Yang XL, et al. Elevated expression of TANK-binding kinase 1 enhances tamoxifen resistance in breast cancer. Proc. Natl. Acad. Sci. U.S.A., 111, E601–E610 (2014).
- 6) Wang Q, Jiang J, Ying GG, et al. Tamoxifen enhances stemness and promotes metastasis of ERα 36+ breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res., 28, 336–358 (2018).
- 7) Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389, 753–758 (1997).
- 8) Wang ZY, Zhang XT, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-α, hER-α 36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. U.S.A., 103, 9063–9068 (2006).
- 9) Shen YY, Zhong J, Liu JH, Liu KH, Zhao J, Xu T, Zeng T, Li ZM, Chen YJ, Ding WJ, Wen GB, Zu XY, Cao RX. Protein arginine N-methyltransferase 2 reverses tamoxifen resistance in breast cancer cells through suppression of ER-alpha 36. Oncol. Rep., 39, 2604–2612 (2018).
- 10) Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst., 96, 926–935 (2004).
- 11) Acconcia F, Barnes CJ, Kumar R. Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology, 147, 1203–1212 (2006).
- 12) Gottardis MM, Robinson SP, Satyaswaroop PG, Jordan VC. Contrasting actions of tamoxifen on endometrial and breast-tumor growth in the athymic mouse. Cancer Res., 48, 812–815 (1988).
- 13) Gottardis MM, Jordan VC. Development of tamoxifen-stimulated growth of MCF-7 tumors in athymic mice after long-term antiestrogen administration. Cancer Res., 48, 5183–5187 (1988).
- 14) Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell, 133, 38–52 (2008).
- 15) Wykosky J, Debinski W. The EphA2 receptor and EphrinA1 ligand in solid tumors: function and therapeutic targeting. Mol. Cancer Res., 6, 1795–1806 (2008).
- 16) Yeddula N, Xia YF, Ke E, Beumer J, Verma IM. Screening for tumor suppressors: loss of ephrin receptor A2 cooperates with oncogenic KRas in promoting lung adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A., 112, E6476–E6485 (2015).
- 17) Wiedemann E, Jellinghaus S, Ende G, Augstein A, Sczech R, Wielockx B, Weinert S, Strasser RH, Poitz DM. Regulation of endothelial migration and proliferation by ephrin-A1. Cell. Signal., 29, 84–95 (2017).
- 18) Hamaoka Y, Negishi M, Katoh H. EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell. Signal., 28, 937–945 (2016).
- 19) Barquilla A, Lamberto I, Noberini R, Heynen-Genel S, Brill LM, Pasquale EB. Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol. Biol. Cell, 27, 2757–2770 (2016).
- 20) Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I, Sakurai H. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat. Commun., 6, 7679 (2015).
- 21) Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang BC. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell, 16, 9–20 (2009).
- 22) Tanaka T, Ozawa T, Oga E, Muraguchi A, Sakurai H. Cisplatin-induced non-canonical endocytosis of EGFR via p38 phosphorylation of the C-terminal region containing Ser-1015 in non-small cell lung cancer cells. Oncol. Lett., 15, 9251–9256 (2018).
- 23) Haryuni R, Tanaka T, Zhou Y, Yokoyama S, Sakurai H. ERK-mediated negative feedback regulation of oncogenic EGFRvIII in glioblastoma cells. Oncol. Lett., 20, 2477–2482 (2020).
- 24) McDonnell DP, Wardell SE. The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr. Opin. Pharmacol., 10, 620–628 (2010).
- 25) Puranik NV, Srivastava P, Bhatt G, Mary D, Limaye AM, Sivaraman J. Determination and analysis of agonist and antagonist potential of naturally occurring flavonoids for estrogen receptor (ER alpha) by various parameters and molecular modelling approach. Sci. Rep., 9, 7450 (2019).
- 26) Pike ACW, Brzozowski AM, Hubbard RE. A structural biologist’s view of the oestrogen receptor. J. Steroid Biochem. Mol. Biol., 74, 261–268 (2000).
- 27) McDonnell DP, Wardell SE, Norris JD. Oral selective estrogen receptor downregulators (SERDs), a breakthrough endocrine therapy for breast cancer. J. Med. Chem., 58, 4883–4887 (2015).
- 28) Friedl A, Jordan VC. Estradiol stimulates growth of estrogen receptor-negative MDA-MB-231 breast-cancer cells in immunodeficient mice by reducing cell loss. Eur. J. Cancer, 30, 1559–1564 (1994).
- 29) Smolen GA, Zhang J, Zubrowski MJ, Edelman EJ, Luo B, Yu M, Ng LW, Scherber CM, Schott BJ, Ramaswamy S, Irimia D, Root DE, Haber DA. A genome-wide RNAi screen identifies multiple RSK-dependent regulators of cell migration. Genes Dev., 24, 2654–2665 (2010).
- 30) Gu WW, Dong N, Wang P, Shi CG, Yang J, Wang J. Tamoxifen resistance and metastasis of human breast cancer cells were mediated by the membrane-associated estrogen receptor ER-alpha 36 signaling in vitro. Cell Biol. Toxicol., 33, 183–195 (2017).
- 31) Walker-Daniels J, Riese DJ 2nd, Kinch MS. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res., 1, 79–87 (2002).
- 32) Sabet O, Stockert R, Xouri G, Bruggemann Y, Stanoev A, Bastiaens PIH. Ubiquitination switches EphA2 vesicular traffic from a continuous safeguard to a finite signalling mode. Nat. Commun., 6, 8047 (2015).
- 33) Gundry C, Marco S, Rainero E, Miller B, Dornier E, Mitchell L, Caswell PT, Campbell AD, Hogeweg A, Sansom OJ, Morton JP, Norman JC. Phosphorylation of Rab-coupling protein by LMTK3 controls Rab14-dependent EphA2 trafficking to promote cell: cell repulsion. Nat. Commun., 8, 14646 (2017).
- 34) Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiainen M, Gstaiger M, Lehti K. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J. Cell Biol., 201, 467–484 (2013).
- 35) Higuchi T, Gohno T, Nagatomo T, Tokiniwa H, Niwa T, Horiguchi J, Oyama T, Takeyoshi I, Hayashi S. Variation in use of estrogen receptor-α gene promoters in breast cancer compared by quantification of promoter-specific messenger RNA. Clin. Breast Cancer, 14, 249–257.e2 (2014).
- 36) Hirata S, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol. Metab., 14, 124–129 (2003).
- 37) Springwald A, Lattrich C, Skrzypczak M, Goerse R, Ortmann O, Treeck O. Identification of novel transcript variants of estrogen receptor alpha, beta and progesterone receptor gene in human endometrium. Endocrine, 37, 415–424 (2010).
- 38) Poola I, Koduri S, Chatra S, Clarke R. Identification of twenty alternatively spliced estrogen receptor alpha mRNAs in breast cancer cell lines and tumors using splice targeted primer approach. J. Steroid Biochem. Mol. Biol., 72, 249–258 (2000).
- 39) Hattori Y, Ishii H, Munetomo A, Watanabe H, Morita A, Sakuma Y, Ozawa H. Human C-terminally truncated ERα variants resulting from the use of alternative exons in the ligand-binding domain. Mol. Cell. Endocrinol., 425, 111–122 (2016).
- 40) Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. U.S.A., 100, 4807–4812 (2003).
- 41) Wang ZY, Zhang XT, Shen P, Loggie BW, Chang YC, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha 36, a novel variant of human estrogen receptor-alpha 66. Biochem. Biophys. Res. Commun., 336, 1023–1027 (2005).
- 42) Schreihofer DA, Duong P, Cunningham RL. N-terminal truncations in sex steroid receptors and rapid steroid actions. Steroids, 133, 15–20 (2018).
- 43) Carmeci C, Thompson DA, Ring HJZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics, 45, 607–617 (1997).
- 44) Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res. Treat., 123, 87–96 (2010).