Abstract
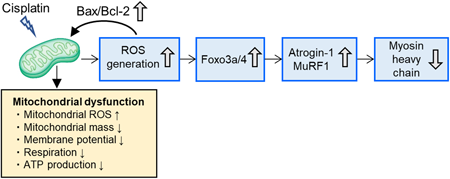
Muscle atrophy is commonly observed during cisplatin chemotherapy, leading to a reduced QOL in cancer patients. Reduced skeletal muscle mass caused by cisplatin treatment results from the activation of ubiquitin ligases–Atrogin-1 and MuRF1, but the precise mechanisms are poorly understood. In this study, we investigated the possible involvement of mitochondrial dysfunction, including reactive oxygen species (ROS) generation and ATP production, in cisplatin-induced muscle atrophy. Skeletal C2C12 myotubes were treated with cisplatin, and gene and protein expression were evaluated. Mitochondrial mass, membrane potential, and ROS levels were measured using fluorescent dyes. Mitochondrial respiratory function, ATP production rates, and glycolytic capacity were also analyzed using an extracellular flux analyzer. Metabolomic analyses were performed using gas chromatography-tandem mass spectrometry. Cisplatin treatment reduced myosin heavy chain expression by activating the ubiquitin-proteasome system. Increased ROS production was observed after cisplatin treatment, followed by significant changes in apoptosis-related gene expression and decrease in mitochondrial mass, membrane potential, respiration, and ATP production. Glycolytic capacity and tricarboxylic acid (TCA) cycle metabolite levels were reduced with cisplatin treatment. Mitochondria-targeted antioxidant mitoquinone mesylate prevented up-regulation of Atrogin-1 gene expression and restored myosin heavy chain levels, accompanied by a decrease in ROS generation, but not mitochondrial ATP production. We concluded that cisplatin-induced myotube atrophy was associated with mitochondrial dysfunction. Reducing ROS generation, rather than promoting ATP production, could be a useful therapeutic strategy for preventing cisplatin-induced muscle atrophy.