Abstract
Macrophages are a key in innate immune responses and play vital roles in homeostasis and inflammatory diseases. Phosphatidylserine-specific phospholipase A1 (PS-PLA1) is a specific phospholipase which hydrolyzes fatty acid from the sn-1 position of phosphatidylserine (PS) to produce lysophosphatidylserine (lysoPS). Both PS and lysoPS are associated with activation of immune cells including macrophages. However, the effect of PS-PLA1 on macrophage inflammation remains unclear. The purpose of this study is to evaluate the role of PS-PLA1 in lipopolysaccharide (LPS)-induced macrophage inflammation. Alterations of PS-PLA1 expression in LPS-stimulated RAW264.7 macrophages were investigated via Western blot. PS-PLA1 stable knockdown and overexpression RAW264.7 cell lines were generated by infecting cells with appropriate lentiviral vectors, respectively. PS-PLA1 expression was found to be dramatically upregulated in RAW264.7 macrophages after LPS stimulation. PS-PLA1 knockdown promotes while PS-PLA1 overexpression ameliorates the release of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and nitric oxide from RAW264.7 cells and M1 macrophage polarization. Additionally, PS-PLA1 knockdown facilitates phosphorylation of p38, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK), while PS-PLA1 overexpression attenuates their phosphorylation. Moreover, mitogen-activated protein kinase (MAPK) inhibitors block the release of TNF-α and IL-1β in PS-PLA1 knockdown RAW264.7 cells after LPS stimulation. These findings suggest PS-PLA1 ameliorates LPS-induced macrophage inflammation by inhibiting MAPKs activation, and PS-PLA1 might be considered as a target for modulating macrophage inflammation.
INTRODUCTION
Macrophages are a key component in innate immunity and play a vital role in homeostasis and inflammatory diseases.1,2) Inflammatory macrophages have a critical role in the clearance of invading pathogens and damaged cells, while uncontrolled inflammation may trigger systemic inflammatory response syndrome or even sepsis.3,4) Thus, it is of great significance to precisely modulate macrophage functions. Macrophages display remarkable plasticity and could be polarized to two main populations with distinct functions, namely the classically activated M1 and alternatively activated M2 subtypes.5) M1 macrophages secrete large amounts of inflammatory factors and display enhanced antigen-presenting ability. They are the primary effector cells for host elimination of pathogens and are associated with immune activation in sepsis. In contrast, M2 macrophages have anti-inflammatory, angiogenic and tissue repair functions and are associated with immune suppression.6,7)
Phosphatidylserine-specific phospholipase A1 (PS-PLA1), also known as phospholipase A1 member A (PLA1A), was first discovered in culture medium of activated rat platelets.8) PS-PLA1 is expressed in various tissues including the liver, lung, prostate gland, and immune cells.9) PS-PLA1, specifically belonging to the pancreatic lipase family, hydrolyzes fatty acid from the sn-1 position of phosphatidylserine (PS) to produce lysophosphatidylserine (lysoPS) and fatty acid. Both PS and lysoPS are associated with the activation of immune cells including macrophages.10) However, abnormal PS-PLA1 expression has been suggested to be linked with autoimmune diseases such as systemic lupus erythematosus,11) lupus nephritis,12) and Graves’ disease13) which indicates potential roles of PS-PLA1 in the regulation of immune responses. The effect of PS-PLA1 on macrophage inflammation remains unclear. Our present study was designed to elucidate the role of PS-PLA1 in lipopolysaccharide (LPS)-induced inflammatory response in macrophages and its underlying mechanisms.
MATERIALS AND METHODS
Cell CultureRAW264.7 cells were obtained from FDCC (Fudan IBS Cell Resource Center, Shanghai, China). Cells were cultured in complete medium of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin (Invitrogen, Karlsruhe, Germany) at 37 °C in 95% air and 5% CO2. Culture medium was replaced every 3–4 d. Subculture was performed when cells covered 80% of the dish.14)
Materials and AntibodiesLPS was bought from Sigma-Aldrich (St. Louis, MO, U.S.A.). PS-PLA1 short hairpin RNA (shRNA) lentivirus and overexpression lentivirus, both expressing green fluorescence protein (GFP), were constructed and packaged by GeneChem (Shanghai, China). Enzyme-linked immunosorbent assay (ELISA) kits used for detection of mouse interleukin (IL)-1β and tumor necrosis factor (TNF)-α were purchased from NeoBioscience (Shenzhen, China). Total nitric oxide (NO) assay kits were acquired from Beyotime (Shanghai, China). Mitogen-activated protein kinases (MAPKs) inhibitors including SB203580 (p38 inhibitor), PD98059 (extracellular signal-regulated kinase (ERK)1/2 inhibitor) and SU3327 (c-Jun N-terminal kinase (JNK) inhibitor) were obtained from MedChemExpress (NJ, U.S.A.). Rabbit antibodies used for Western blot including anti-p-ERK1/2, anti-ERK1/2, anti-p-p38, anti-p38, anti-p-JNK and anti-JNK were purchased from Cell Signaling Technology (Danvers, MA, U.S.A.). A rabbit anti-β-αctin antibody was purchased from Abways Technology (Shanghai, China). A mouse anti-PS-PLA1 antibody was purchased from Abcam (Cambridge, U.K.). Antibodies used for flow cytometry including a CD86 (B7-2) monoclonal antibody and a CD206 (MMR) monoclonal antibody were purchased from eBioscience (San Diego, CA, U.S.A.).
Gene Knockdown and OverexpressionThree sets of shRNAs against PS-PLA1 were designed and packaged into lentivirus expressing GFP by GeneChem (Shanghai, China) while the efficiencies of PS-PLA1 shRNAs were initially tested. The shRNA with the highest PS-PLA1 knockdown efficiency was used for further experiments. For overexpression of PS-PLA1 in RAW264.7 macrophages, the cells were transfected with the lentivirus expressing GFP and contained the mouse PS-PLA1 sequences (GeneChem, Shanghai, China) according to the manufacturer’s instructions.
ELISACulture supernatants of RAW264.7 macrophages were collected and then centrifuged at 4 °C to remove particles. Levels of IL-1β and TNF-α in cell supernatant were detected by commercially available ELISA kits according to the manufacturer’s instructions (NeoBioscience, Shenzhen, China). The value of optical density (OD) of each well was measured by a microplate reader at the wavelength of 450 nm, while standard curves were used for quantifying the concentrations of IL-1β and TNF-α in the samples, respectively.
Nitrite Colorimetric AssayCell supernatants were collected and then centrifuged at 4 °C. Concentrations of NO were detected by the nitrite colorimetric method using commercially available total NO assay kit (Beyotime, Shanghai, China) following the manufacturer’s instructions.
Flow Cytometric AssayCD86 was used to mark M1 macrophages, while CD206 was used to identify M2 macrophages. The cells were collected. The Fc receptors were blocked using the CD16/CD32 monoclonal antibodies. That was followed by the incubation with CD86 antibodies at room temperature. Next, the Fix/Perm Buffer was used for membrane breaking. Perm/Wash Buffer was added for washing after which the CD206 antibodies were added for incubation. Following this, the samples were detected using a BD FACSCanto II flow cytometer. Lastly, the data were analyzed by BD FACSDiva software.
Protein–Protein Interaction Network AnalysisProtein–protein interaction information was acquired from the STRING database (http://www.string-db.org/),15) which aims to integrate all known and predicted associations between proteins, including both physical interactions as well as functional associations.16) “PS-PLA1” was used as the query protein name, “Homo sapiens” was selected and “no more than 20 interactors” was chosen. The queried results were used for reference in next experiments.
Western Blot AnalysisTotal cellular proteins were extracted using ice-cold radio immunoprecipitation assay (RIPA) lysis buffer (KeyGEN BioTECH, Nanjing, China) with the addition of protease inhibitors and phosphatase inhibitors.17,18) Protein quantification was performed using a BCA Kit (KeyGEN BioTECH, Nanjing, China). Proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then electrotransferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with Tris Buffered Saline with Tween 20 (TBST) buffer containing 5% skim milk or 5% (w/v) bovine serum albumin (BSA) (for phosphorylated proteins), followed by incubation with primary antibodies overnight at 4 °C. After incubation with secondary antibodies for 1 h at room temperature, chemiluminescence detection of the bands was performed in ChemiScope Series scanning system (Clinx Science, Shanghai, China).
Statistical AnalysisData were expressed as mean ± standard deviation (S.D.). One-way ANOVA was used for comparisons among groups, and differences were considered statistically significant at p < 0.05.
RESULTS
LPS Stimulation Increases PS-PLA1 Expression in RAW264.7 MacrophagesTo investigate whether PS-PLA1 participates in LPS-induced inflammatory responses in macrophages, the expression of PS-PLA1 in RAW264.7 macrophages after LPS stimulation was measured by Western blot. The results showed that the expression level of PS-PLA1 increased significantly after LPS stimulation. It reached the peak at 16 h (Fig. 1), suggesting a possible role of PS-PLA1 in LPS-induced macrophage inflammation.
PS-PLA1 Knockdown Promotes the Release of Inflammatory Mediators from RAW264.7 MacrophagesTo further clarify the role of PS-PLA1 in LPS-induced macrophage inflammation, a shRNA-mediated PS-PLA1 stable knockdown RAW264.7 cell line was generated (Supplementary Fig. 1). The levels of inflammatory mediators including TNF-α, IL-1β and NO were dramatically increased in the supernatants of RAW264.7 cells under both basal state and LPS treatment (Figs. 2A–C). This finding suggests that PS-PLA1 knockdown promotes LPS-induced inflammatory responses in macrophages.
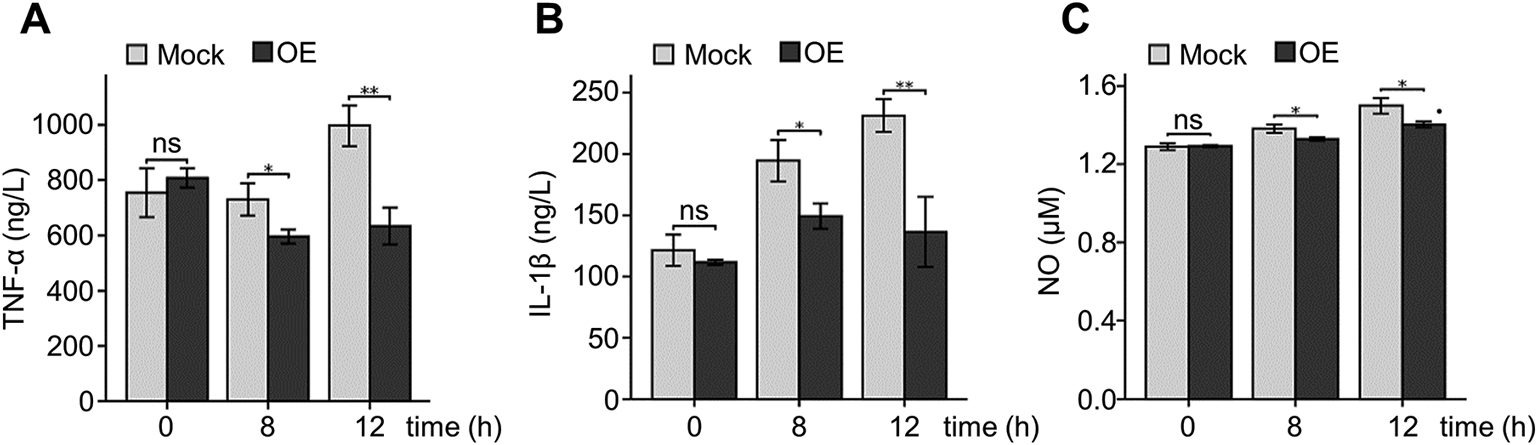
PS-PLA1 Overexpression Ameliorates the Release of Inflammatory Mediators from LPS-Treated RAW264.7 MacrophagesThe effects of PS-PLA1 overexpression on LPS-induced release of inflammatory mediators were also investigated. A PS-PLA1 stable overexpression RAW264.7 cell line was established (Supplementary Fig. 2). The expression levels of TNF-α, IL-1β and NO were significantly decreased in the overexpression (OE) group after LPS stimulation (Figs. 3A–C). This finding suggests that overexpression of PS-PLA1 inhibits LPS-induced inflammatory response in macrophages.
PS-PLA1 Knockdown Promotes M1 While Inhibits M2 Polarization in RAW264.7 MacrophagesMacrophages acquire a polarized phenotype as either pro-inflammatory M1 macrophages or anti-inflammatory M2 macrophages in response to microenvironmental stimulations. Therefore, the focus of this study was to examine the effect of PS-PLA1 knockdown on macrophage polarizations via flow cytometry. The results indicate that PS-PLA1 knockdown promotes M1 polarization while inhibiting M2 polarization in RAW264.7 macrophages (Figs. 4A, B).
PS-PLA1 Overexpression Inhibits M1 but Promotes M2 Polarization in RAW264.7 MacrophagesNext, the role of PS-PLA1 overexpression in RAW264.7 macrophage polarizations was investigated via flow cytometry. In line with previous results, it was found that PS-PLA1 overexpression inhibits the M1 polarization but promotes M2 polarization in RAW264.7 macrophages (Figs. 5A, B).
Involvement of MAPKs in PS-PLA1 Regulated LPS-Induced Macrophage ResponsesThe underlying mechanisms by which PS-PLA1 regulates LPS-induced inflammation and polarization in RAW264.7 cells were further investigated. By looking in the STRING database (http://string-db.org), a useful tool for predicting functional associations between proteins, we found that the interaction between PS-PLA1 and MAPK1/MAPK3 has been suggested (Supplementary Fig. 3). More importantly, MAPKs are also the downstream of LPS/Toll-like receptor 4 (TLR4) signaling. Therefore, alterations in MAPKs, including p38, ERK1/2 and JNK after PS-PLA1 knockdown or overexpression were examined. The Western blot results showed the phosphorylation of these MAPKs significantly increases in PS-PLA1 knockdown RAW264.7 cells after LPS stimulation (Fig. 6A), while PS-PLA1 overexpression inhibits the phosphorylation of these MAPKs (Fig. 6B), indicating a negatively regulatory effect of PS-PLA1 on the phosphorylation of MAPKs. Next, MAPKs in PS-PLA1 knockdown RAW264.7 cells were inhibited by adding the inhibitors of p38, ERK and JNK. ELISA results showed significant reduction in LPS-induced release of TNF-α (Fig. 6C) and IL-1β (Fig. 6D). This indicates that PS-PLA1 attenuates LPS-induced macrophage inflammation by inhibiting MAPKs activation.
DISCUSSION
In this study, it was discovered that PS-PLA1 attenuates LPS-induced macrophage inflammation by inhibiting MAPKs activation. PS-PLA1 knockdown promotes while PS-PLA1 overexpression ameliorates the LPS-induced release of inflammatory mediators from macrophages and macrophage M1 polarization. It was mechanistically determined that PS-PLA1 inhibits the phosphorylation of MAPKs including p38, ERK and JNK.
PS-PLA1 is a widely distributed phospholipase in the body and acts specifically on PS to generate lysoPS,19,20) while lysoPS is thought to be an immunological regulator.21) To evaluate whether PS-PLA1 is involved in macrophage inflammation, PS-PLA1 expression was examined in LPS-stimulated RAW264.7 macrophages. This study found that PS-PLA1 expression was significantly upregulated. Previous studies have also reported that elevated PS-PLA1 level is closely related to autoimmune diseases including systemic lupus erythematosus,11) lupus nephritis,12) and Graves’ disease.13) A prior study used various Toll-like receptor ligands including heat-killed Listeria monocytogenes (HKLM), polyinosine-polycytidylic acid (poly(I:C)), LPS, flagellin, loxoribine and ODN2006 to stimulate THP-1-derived macrophages. The study found that LPS induces increased expression of PS-PLA1.10) Another study using microarray gene analysis found that LPS stimulation induces a 24.3-fold increase in PS-PLA1 precursor expression in rat liver tissue.22) Findings in this study are consistent with these previously published literatures, suggesting that PS-PLA1 expression is upregulated after LPS treatment and it may play an important role in inflammatory responses. In addition, PS-PLA1 knockdown and overexpression RAW264.7 cells were generated and the effects of PS-PLA1 on LPS-induced macrophage inflammation were examined using these cells. We found that PS-PLA1 knockdown promotes while PS-PLA1 overexpression attenuates LPS-induced macrophage inflammation. Despite previous studies’ reports suggesting increased PS-PLA1 expression in autoimmune diseases and considering PS-PLA1 as a diagnostic marker,11–13) the role of PS-PLA1 in inflammation is not clear yet. Our present work has proved the inhibitory effects of PS-PLA1 on LPS-induced TNFα and IL-1β production and therefore PS-PLA1 might serve as a novel inflammatory inhibitor.
The underlying mechanisms by which PS-PLA1 negatively regulated LPS-induced macrophage inflammation were explored and MAPKs were found to be involved. LPS, the ligand for TLR4, has been known to induce signal transduction via TLR4,23) while the downstream of TLR4 signaling are nuclear factor-kappaB (NF-κB), MAPKs, and interferon regulatory factor 3 (IRF3).24) The pivotal role of MAPK pathway in macrophage inflammation has also been supported by other reports.25–28) MAPKs, comprising of ERK, p38 and JNK, have controlled the mechanism of LPS-stimulated inflammatory responses in macrophages.29) However, the effects of PS-PLA1 in MAPKs activation have not been investigated yet. This study showed that PS-PLA1 knockdown promotes while PS-PLA1 overexpression alleviates the phosphorylation of p38, ERK and JNK which are three important members of the mammalian MAPK superfamily.30) The results of this study are supported by the STRING database which aims to provide a critical assessment and integration of protein–protein interactions.31) The STRING database also suggests the interaction of PS-PLA1 with MAPK1/MAPK3. However, one limitation of our present work is that we have not further clarified whether this negative regulation of PS-PLA1 on MAPKs is achieved by direct physical interactions or indirect effects through lysoPS, because the PS-PLA1-produced lysoPS has been reported to suppress LPS signaling.32) This issue needs to be addressed in future studies.
To demonstrate the enhanced inflammatory response of PS-PLA1 knockdown macrophages was resulted from MAPKs activation, SB203580, SU3327 and PD98059 were added to inhibit the activation of p38, JNK and ERK, respectively. It was discovered that inhibition of MAPK partially blocks the promotive effect of PS-PLA1 knockdown on LPS-induced release of TNF-α and IL-1β from RAW264.7 macrophages. Our results are similar to previous works which show that costunolide inhibits IL-1β expression by inhibiting MAPK activity in LPS-stimulated RAW 264.7 cells33) and extract from Grateloupia lanceolata inhibits LPS-induced production of NO and IL-1β via down-regulation of MAPK in macrophages.27) However, it appears that MAPKs are not isolated molecules; rather, they function as an interactive network with alternative pathways or feedback mechanisms.34,35) Therefore, the inhibition of p38, JNK or ERK alone cannot completely block the PS-PLA1 knockdown-induced pro-inflammatory effects of macrophages. This finding is consistent with previous clinical studies on p38 inhibitors for rheumatoid arthritis which showed that inhibition of p38 resulted in only a transient decrease in the level of serum C-reactive protein.36) It indicates that other pathways might have alternative effects.
In conclusion, this study revealed that PS-PLA1 alleviates LPS-induced macrophage inflammation by inhibiting MAPKs activation (Fig. 7), and PS-PLA1 might be considered as a potential target for modulating macrophage inflammation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82172182 and 82102311), National Natural Science Foundation of Jiangsu Province (BK20211136 and BK20190247), China Postdoctoral Science Foundation (2020M683718).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Liu F, Qiu H, Xue M, Zhang S, Zhang X, Xu J, Chen J, Yang Y, Xie J. MSC-secreted TGF-β regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res. Ther., 10, 345 (2019).
- 2) Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity, 32, 593–604 (2010).
- 3) Ma J, Wei K, Liu J, Tang K, Zhang H, Zhu L, Chen J, Li F, Xu P, Liu J, Fang H, Tang L, Wang D, Zeng L, Sun W, Xie J, Liu Y, Huang B. Glycogen metabolism regulates macrophage-mediated acute inflammatory responses. Nat. Commun., 11, 1769 (2020).
- 4) Burton DGA, Stolzing A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev., 43, 17–25 (2018).
- 5) Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol., 8, 958–969 (2008).
- 6) Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front. Immunol., 10, 1462 (2019).
- 7) Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis, 17, 109–118 (2014).
- 8) Sato T, Aoki J, Nagai Y, Dohmae N, Takio K, Doi T, Arai H, Inoue K. Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family. J. Biol. Chem., 272, 2192–2198 (1997).
- 9) Zhao Y, Hasse S, Bourgoin SG. Phosphatidylserine-specific phospholipase A1: A friend or the devil in disguise. Prog. Lipid Res., 83, 101112 (2021).
- 10) Hosono H, Homma M, Ogasawara Y, Makide K, Aoki J, Niwata H, Watanabe M, Inoue K, Ohkohchi N, Kohda Y. Expression of phosphatidylserine-specific phospholipase A(1) mRNA in human THP-1-derived macrophages. Cell Transplant., 19, 759–764 (2010).
- 11) Sawada T, Kurano M, Shirai H, Iwasaki Y, Tahara K, Hayashi H, Igarashi K, Fujio K, Aoki J, Yatomi Y. Serum phosphatidylserine-specific phospholipase A1 as a novel biomarker for monitoring systemic lupus erythematosus disease activity. Int. J. Rheum. Dis., 22, 2059–2066 (2019).
- 12) Iwata Y, Kitajima S, Yamahana J, Shimomura S, Yoneda-Nakagawa S, Sakai N, Furuichi K, Ogura H, Sato K, Toyama T, Yamamura Y, Miyagawa T, Hara A, Shimizu M, Ohkawa R, Kurano M, Yatomi Y, Wada T. Higher serum levels of autotaxin and phosphatidylserine-specific phospholipase A1 in patients with lupus nephritis. Int. J. Rheum. Dis., 24, 231–239 (2021).
- 13) Nakawatari K, Kurano M, Araki O, Nishikawa M, Shimamoto S, Igarashi K, Aoki J, Murakami M, Yatomi Y. Elevated phosphatidylserine-specific phospholipase A1 level in hyperthyroidism. Clin. Chim. Acta, 503, 99–106 (2020).
- 14) Liu C, Hu YH, Han Y, Wang YB, Zhang Y, Zhang XQ, He DF, Ren HM, Liu YK, Wang HY, Tan T, Lin PH, Li HC, Rovin BH, Ma JJ, Zeng CY. MG53 protects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis. Acta Pharmacol. Sin., 41, 1457–1464 (2020).
- 15) Chengyu Y, Long Z, Bin Z, Hong L, Xuefei S, Congjuan L, Caixia C, Yan X. Linarin protects the kidney against ischemia/reperfusion injury via the inhibition of bioactive ETS2/IL-12. Biol. Pharm. Bull., 44, 25–31 (2021).
- 16) Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res., 49 (D1), D605–D612 (2021).
- 17) Liu C, Chen K, Wang H, Zhang Y, Duan X, Xue Y, He H, Huang Y, Chen Z, Ren H, Wang H, Zeng C. Gastrin attenuates renal ischemia/reperfusion injury by a PI3K/Akt/Bad-mediated anti-apoptosis signaling. Front. Pharmacol., 11, 540479 (2020).
- 18) Liu C, Li X, Fu J, Chen K, Liao Q, Wang J, Chen C, Luo H, Jose PA, Yang Y, Yang J, Zeng C. Increased AT1 receptor expression mediates vasoconstriction leading to hypertension in Snx1−/− mice. Hypertens. Res., 44, 906–917 (2021).
- 19) Uranbileg B, Kurano M, Sato M, Ikeda H, Ishizawa T, Hasegawa K, Kokudo N, Yatomi Y. Possible involvement of PS-PLA1 and lysophosphatidylserine receptor (LPS1) in hepatocellular carcinoma. Sci. Rep., 10, 2659 (2020).
- 20) Kurano M, Miyagaki T, Miyagawa T, Igarashi K, Shimamoto S, Ikeda H, Aoki J, Sato S, Yatomi Y. Association between serum autotaxin or phosphatidylserine-specific phospholipase A1 levels and melanoma. J. Dermatol., 45, 571–579 (2018).
- 21) Sugo T, Tachimoto H, Chikatsu T, Murakami Y, Kikukawa Y, Sato S, Kikuchi K, Nagi T, Harada M, Ogi K, Ebisawa M, Mori M. Identification of a lysophosphatidylserine receptor on mast cells. Biochem. Biophys. Res. Commun., 341, 1078–1087 (2006).
- 22) Deaciuc IV, Peng X, D’Souza NB, Shedlofsky SI, Burikhanov R, Voskresensky IV, de Villiers WJ. Microarray gene analysis of the liver in a rat model of chronic, voluntary alcohol intake. Alcohol, 32, 113–127 (2004).
- 23) Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem., 274, 10689–10692 (1999).
- 24) Sweet CR, Conlon J, Golenbock DT, Goguen J, Silverman N. YopJ targets TRAF proteins to inhibit TLR-mediated NF-kappaB, MAPK and IRF3 signal transduction. Cell. Microbiol., 9, 2700–2715 (2007).
- 25) Cha SM, Cha JD, Jang EJ, Kim GU, Lee KY. Sophoraflavanone G prevents Streptococcus mutans surface antigen I/II-induced production of NO and PGE2 by inhibiting MAPK-mediated pathways in RAW 264.7 macrophages. Arch. Oral Biol., 68, 97–104 (2016).
- 26) Han BH, Lee YJ, Yoon JJ, Choi ES, Namgung S, Jin XJ, Jeong DH, Kang DG, Lee HS. Hwangryunhaedoktang exerts anti-inflammation on LPS-induced NO production by suppressing MAPK and NF- κB activation in RAW264.7 macrophages. J. Integr. Med., 15, 326–336 (2017).
- 27) Kim DH, Kim ME, Lee JS. Inhibitory effects of extract from G. lanceolata on LPS-induced production of nitric oxide and IL-1β via down-regulation of MAPK in macrophages. Appl. Biochem. Biotechnol., 175, 657–665 (2015).
- 28) Niu X, Wang Y, Li W, Zhang H, Wang X, Mu Q, He Z, Yao H. Esculin exhibited anti-inflammatory activities in vivo and regulated TNF-α and IL-6 production in LPS-stimulated mouse peritoneal macrophages in vitro through MAPK pathway. Int. Immunopharmacol., 29, 779–786 (2015).
- 29) Sukketsiri W, Tanasawet S, Moolsap F, Tantisira MH, Hutamekalin P, Tipmanee V. ECa 233 Suppresses LPS-induced proinflammatory responses in macrophages via suppressing ERK1/2, p38 MAPK and Akt pathways. Biol. Pharm. Bull., 42, 1358–1365 (2019).
- 30) Yu J, Sun X, Goie JYG, Zhang Y. Regulation of host immune responses against influenza A virus infection by mitogen-activated protein kinases (MAPKs). Microorganisms, 8, 1067 (2020).
- 31) Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res., 43 (D1), D447–D452 (2015).
- 32) Nishikawa M, Kurano M, Ikeda H, Aoki J, Yatomi Y. Lysophosphatidylserine has bilateral effects on macrophages in the pathogenesis of atherosclerosis. J. Atheroscler. Thromb., 22, 518–526 (2015).
- 33) Kang JS, Yoon YD, Lee KH, Park SK, Kim HM. Costunolide inhibits interleukin-1beta expression by down-regulation of AP-1 and MAPK activity in LPS-stimulated RAW 264.7 cells. Biochem. Biophys. Res. Commun., 313, 171–177 (2004).
- 34) Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J., 22, 954–965 (2008).
- 35) Asthagiri AR, Lauffenburger DA. A computational study of feedback effects on signal dynamics in a mitogen-activated protein kinase (MAPK) pathway model. Biotechnol. Prog., 17, 227–239 (2001).
- 36) Paunovic V, Harnett MM. Mitogen-activated protein kinases as therapeutic targets for rheumatoid arthritis. Drugs, 73, 101–115 (2013).