Abstract
Despite advances in colorectal cancer (CRC) treatment, most advanced CRC patients who experience disease progression after chemotherapy, targeted therapy, and immunotherapy face a situation in which there is no available medicine. Thus, new therapeutic drugs for CRC are urgently needed. Studies have shown that cholesteryl ester transfer protein (CETP) has a vital role in tumor development and is a possible target for CRC therapy. We found that Evacetrapib, a CETP inhibitor, suppressed CRC cell growth by inhibiting the Wnt/β-catenin signaling pathway and activating the c-Jun NH2-terminal kinase (JNK) signaling pathway in CRC. Therefore, Evacetrapib displays an anti-cancer effect and is a possible option for treating CRC.
INTRODUCTION
Globally, colorectal cancer (CRC) is the 3rd most prevalent tumor and the 2nd largest cause of tumor-associated deaths.1) As a result of the insidious onset of CRC, most cases are diagnosed in the late stages.2) The main treatments for advanced CRC are chemotherapy, targeted therapy and immunotherapy.3) However, the efficacy of many chemotherapeutic agents and targeted therapeutic drugs is often unsatisfactory because of drug resistance and side effects.4) Moreover, patients with microsatellite stable (MSS) CRC do not benefit from immunotherapy.5,6) Thus, it is vital to develop new drugs for CRC treatment.
Cholesteryl ester transfer protein (CETP), a plasma glycoprotein, is involved in the cholesterol reverse transport (RCT) pathway by facilitating cholesterol esters translocation from high-density lipoprotein (HDL) to very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), and low-density lipoprotein (LDL).7) CETP predisposes individuals to cardiovascular diseases. Recent studies have demonstrated that CETP shows increased activity in CRC, indicating that inhibiting CETP may be a potential approach to treating CRC.8,9) Evacetrapib selectively inhibits CETP and reduces cardiovascular events by enhancing high-density lipoprotein cholesterol (HDL-C) levels.10) Nevertheless, the significance of Evacetrapib in CRC is yet to be fully determined. We found that Evacetrapib could inhibit CRC cell growth by initiating cell proliferation inhibition and apoptosis. Additionally, suppressing the Wnt/β-catenin signaling pathway and activating the c-Jun NH2-terminal kinase (JNK) signaling pathway were involved in the role of Evacetrapib in CRC. These data strongly suggest that Evacetrapib may be a new optional drug for treating CRC.
MATERIALS AND METHODS
ReagentsEvacetrapib (APExBIO, Houston, TX, U.S.A.), SP600125 (Sigma-Aldrich, St. Louis, MO, U.S.A.) and z-VAD-fmk (Selleck Chemicals, Houston, TX, U.S.A.) were resuspended in dimethyl sulfoxide (DMSO) and kept at −20 °C. P21 Waf1/Cip1, caspase3, poly(ADP-ribose)polymerase (PARP), cleaved-caspase3, survivin, Bcl-xl, phospho-β-catenin (Ser675), stress-activated protein kinase (SAPK)/JNK, X-linked inhibitor of apoptosis (XIAP), and phospho-SAPK/JNK (Thr183/Tyr185) antibodies were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.). β-Catenin was procured from Abcam (Cambridge, Cambridgeshire, U.K.). β-Actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), anti-mouse as well as anti-rabbit immunoglobulin G horseradish peroxidase-conjugated secondary antibodies were procured from the Proteintech Group (Chicago, IL, U.S.A.).
Cell CultureCRC cell lines HCT116, HCT8, SW620, DLD1, LoVo, as well as RKO were acquired from the Culture Collection of the Chinese Academy of Science (Shanghai, China). Cells were grown in 10% fetal bovine serum-supplemented RPMI 1640 medium (Gibco Life Technologies, Carlsbad, CA, U.S.A.) with 100 U/mL and 10 µg/mL penicillin and streptomycin, respectively. Culture was done in a 5% CO2 environment at 37 °C.
Assessment of Cell ViabilityA total of 1 × 104 cells per well were inoculated into 96-well plates and exposed to different Evacetrapib doses for 48 h. Then, 20 µL Cell Counting Kit-8 (CCK-8) (KeyGEN, Nanjing, Jiangsu, China) reagent was supplemented to the wells followed by incubation. Optical densities were evaluated by a Varioskan Flash multimode reader (Thermo Fisher Scientific, Waltham, MA, U.S.A.).
5-Bromo-2′-deoxyuridine (BrdU)-Labeling ExperimentsA total of 5 × 105 cells in RPMI 1640 medium were inoculated in 6-well plates. After incubation with varying doses of Evacetrapib for 24 h, cells were evaluated using a fluoroisothiocyanate (FITC)-BrdU cell proliferation kit (KeyGEN, Nanjing, Jiangsu, China), as instructed by the manufacturer. The role of evacetrapib in cell proliferation was assessed by CytoFLEX (Beckman Coulter, CA, U.S.A.).
Colony Formation AssayHCT116 as well as DLD1 cells were inocluated into a 6-well plate (1 × 103 cells/well) and exposed to varying doses of Evacetrapib for 7 d. Then, they were washed using phosphate buffered saline (PBS), fixed for 5 min in anhydrous methanol followed by 5 min of staining with crystal violet (0.5%) at room temperature (r.t.). Colony images were captured by an Epson scanner (Suwa, Nagano, Japan).
Cell Cycle EvaluationIncubation of HCT116 as well as DLD1 cells was done with Evacetrapib for 24 h and thereafter fixed overnight in ethanol at 4 °C. They were washed using PBS, labeled using propidium iodide (PI, BD Biosciences, Franklin Lake, NJ, U.S.A.) and analyzed by CytoFLEX.
Cell Apoptosis AnalysisIncubation of HCT116 and DLD1 cells was done in the presence of increasing Evacetrapib doses for designated times. Apoptosis was evaluated by an Annexin V FITC/PI double staining kit (KeyGEN, Nanjing, Jiangsu, China) after Evacetrapib treatment. Briefly, cells were obtained, washed, and thereafter stained with a binding buffer (500 µL), which included Annexin V-FITC (5 µL) and PI (5 µL), in the dark at r.t. for 15 min. In addition, apoptosis was determined by CytoFLEX.
Wnt Luciferase Activity AssayHCT116 cells transfected with pGL4.49 and pRL-TK plasmids were inoculated in 96-well plates, incubated for 24 h and treated with Evacetrapib. Luciferase activities were evaluated using a Luciferase Assay Kit (Promega, Madison, WI, U.S.A.), as instructed by the manufacturer. Measurements were performed by a Thermo Scientific Varioskan Flash Multimode Reader (Thermo Fisher Scientific, Waltham, MA, U.S.A.).
Real-Time PCRTotal RNA from cells were extracted by an RNA Quick Purification kit (ESscience, Shanghai, China). Then, we used the PrimeScript RT reagent Kit (TaKaRa, Dalian, Liaoning, China) for reverse transcribing RNA to cDNA. The SYBR Premix Ex Taq II kit (TaKaRa, Dalian, Liaoning, China) was used for real-time PCR on an ABI 7500 system (ABI, New York, NY, U.S.A.). Gene expressions were evaluated via the ΔΔCt method. Primer sequences in this assay were: Axin2 forward: 5′-CTGGCTTTGGTGAACTGTTG-3′, Axin2 reverse: 5′-AGTTGCTCACAGCCAAGACA-3′; CyclinD1 forward: 5′-GCTGCGAAGTGGAAACCATC-3′, CyclinD1 reverse: 5′-CCTCCTTCTGCACACATTTGAA-3′; c-Myc forward: 5′-GGCTCCTGGCAAAAGGTCA-3′, c-Myc reverse: 5′-CTGCGTAGTTGTGCTGATGT-3′; Survivin forward: 5′-AGGACCACCGCATCTCTACAT-3′, Survivin reverse: 5′-AAGTCTGGCTCGTTCTCAGTG-3′; GAPDH forward: 5′-GCACCGTCAAGGCTGAGAAC-3′, GAPDH reverse: 5′-TGGTGAAGACGCCAGTGGA-3′.
Western Blot AssayAfter the cells had been lysed in the radio immunoprecipitation assay (RIPA) lysis buffer (Cell Signaling Technology, Beverly, MA, U.S.A.) with phosphatase and protease inhibitors (KeyGEN, Nanjing, Jiangsu, China), a bicinchoninic acid (BCA) protein assay kit (Invitrogen, Carlsbad, CA, U.S.A.) was used for protein quantification. Equal protein amounts were separated by sodium dodecyl sulfate- polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis, transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Anaheim, CA, U.S.A.), blocked with 5% milk, followed by incubation in the presence of corresponding primary antibodies for 4 °C overnight. The membrane was washed using TBST followed by incubation at r.t. for 1 h with secondary antibodies. Enhanced chemiluminescence (ECL) (Bio-Rad, Hercules, CA, U.S.A.) was used to detect signals.
RESULTS
Evacetrapib Suppresses the Proliferation of CRC CellsFigure 1A shows that CRC cell viability was suppressed with increasing Evacetrapib doses, indicating that Evacetrapib has anticancer properties on CRC cells. Human CRC cell lines (DLD1 and HCT116) were selected for further studies. BrdU, an analog of thymine, was used as a marker of cell proliferation.11) Thus, BrdU staining was performed to evaluate the effects of Evacetrapib on cell proliferation. To further evaluate the long-term effects of Evacetrapib on CRC cell growth, we adopted the clone formation assay. The BrdU labeling experiment and clone formation analysis verified that Evacetrapib significantly inhibited cell proliferation (Figs. 1B, C). Interestingly, the results of clone formation assay showed that DLD1 was more sensitive to evacetrapib than HCT116. This is in inconsistent with the experimental results of CCK-8 and BrdU. Clone formation is associated with cell stemness,12,13) and the spheroidizing ability of DLD1 is significantly weaker than that of HCT116.14) Therefore, this inconsistency may be caused by different cell stemness. Furthermore, we explored the role of Evacetrapib on the cell cycle distribution (Fig. 1D). After 24 h of Evacetrapib treatment, an increased abundance of cells in the G1/S phase was noted, implying that Evacetrapib initiates CRC cell cycle arrest within the G1/S phase. P21 Waf1/Cip1 is the most representative negative regulator of the G1/S transition and thus contributes to “G1 arrest.”15) Western blot assay revealed that after Evacetrapib treatment, p21 protein levels were increased (Fig. 1E). These findings confirmed that Evacetrapib inhibits CRC cell proliferation in vitro.
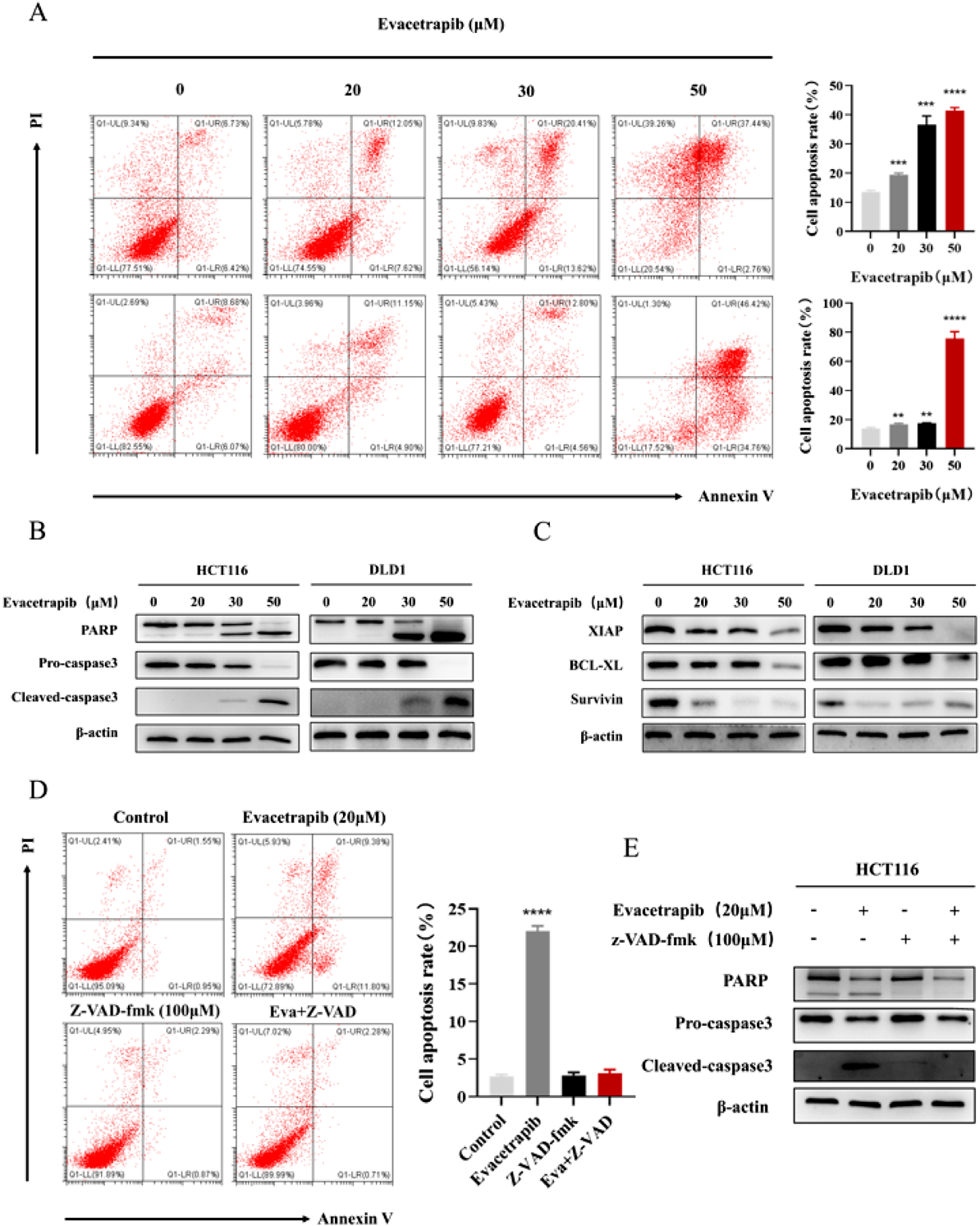
Evacetrapib Initiates CRC Cell ApoptosisResistance to cell death is one of the characteristics of tumors.16) To explore whether Evacetrapib can initiate CRC cell apoptosis, annexin V-FITC/PI staining and flow cytometry were done to assess the percentage of apoptotic cells. After treatment with Evacetrapib, an increase in the abundance of apoptotic cells was observed (Fig. 2A). This result was ultimately confirmed following the detection of the expression of apoptosis-related proteins. Cleaved PARP is considered one of the markers for apoptosis.17) When cell apoptosis occurs, the expression of cleaved PARP increases, but PARP decreases. Moreover, caspase3, the final apoptosis executive molecule of the endogenous mitochondrial cytochrome release pathway and the exogenous death receptor pathway, was activated.18) The results are presented in Fig. 2B. Further, we assessed the levels of BCL-2 and IAP family proteins. After Evacetrapib treatment, levels of antiapoptotic proteins XIAP, survivin, as well as Bcl-xl were suppressed (Fig. 2C). Lastly, in order to confirm that Evacetrapib relies on caspase to induce apoptosis, we treated cells with caspase inhibitor Z-VAD-fmk and Evacetrapib. Results are shown in Figs. 2D and E, the caspase inhibitor Z-VAD-fmk could reverse the apoptosis-inducing activity of evacetrapib by flow cytometry and Western blot assay, indicating that Evacetrapib induced apoptosis depends on caspase.
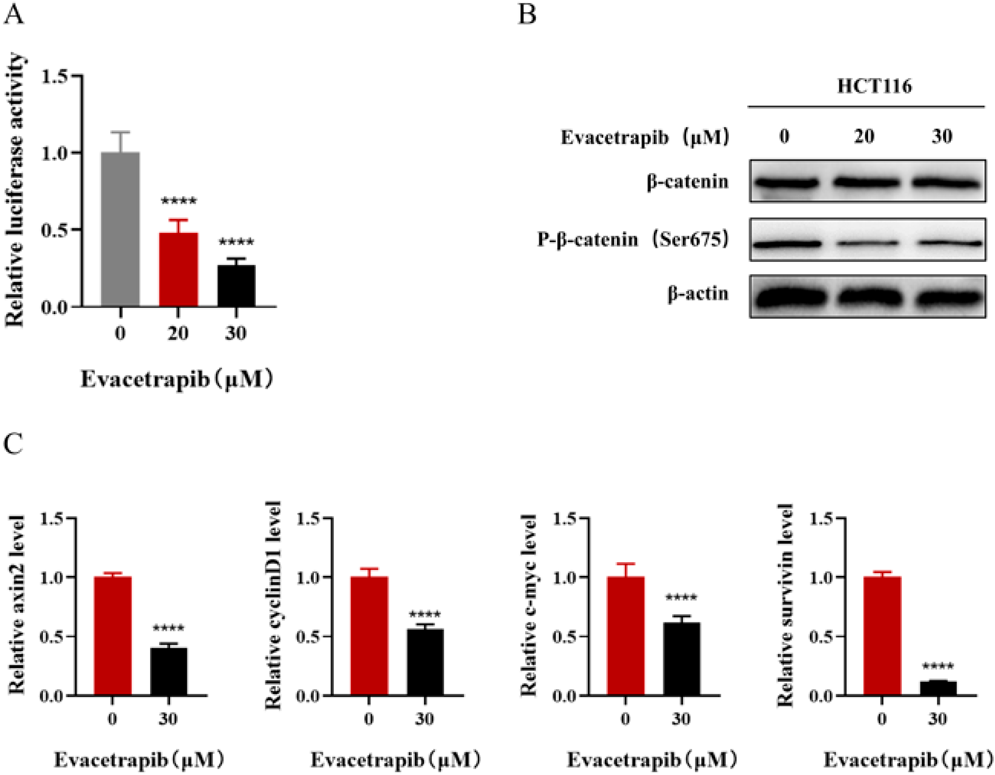
Evacetrapib Inhibits the Wnt/β-Catenin Signaling Pathway in CRCTo evaluate the effects of Evacetrapib on the Wnt/β-catenin signaling pathway in CRC, we transfected the Wnt luciferase reporter plasmid pGL4.49 into HCT116 cells. Evacetrapib dose-dependently inhibited the activity of luciferase (Fig. 3A). Furthermore, phosphorylation of β-catenin Ser675 (p-β-catenin (Ser675)), as well as total β-catenin were assessed by Western blot assay. Figure 3B shows that Evacetrapib treatment did not show any apparent changes in total p-β-catenin levels but markedly decreased p-β-catenin (Ser675) levels in HCT116 cells. Simultaneously, mRNA levels of target genes involved in Wnt signaling, including axin2, cyclinD1, c-myc, and survivin, were analyzed. Evacetrapib inhibited the levels of the above genes (Fig. 3C). These findings revealed that inhibition of the Wnt/β-catenin signaling pathway was associated with the role of Evacetrapib in CRC.
Evacetrapib Induces CRC Cell Apoptosis by Activating the JNK Signaling PathwayInvolvement of the JNK signaling pathway in CRC apoptosis is well known.19) Therefore, we hypothesized that Evacetrapib initiates CRC cell apoptosis by activating JNK. First, we used dual phospho-specific JNK antibodies to analyze Evacetrapib-mediated alterations in JNK phosphorylation through a Western blot assay. The phospho-JNK (p-JNK) levels were elevated in HCT116 cells incubated with Evacetrapib, while the total JNK levels remained unchanged (Fig. 4A). Furthermore, to confirm whether the JNK signaling pathway has a role in Evacetrapib-mediated HCT116 cell apoptosis, cells were cotreated with SP600125, a JNK inhibitor. Suppression of JNK by SP600125 decreased the p-JNK level (Fig. 4B). Flow cytometry and Western blot assay showed that SP600125 dramatically reversed Evacetrapib-mediated apoptosis (Figs. 4C, D). These findings implied that Evacetrapib induces CRC cell apoptosis by activating the JNK signaling pathway.
DISCUSSION
CRC, a common malignant carcinoma of the digestive tract, is usually diagnosed in advanced stages and has a low five-year survival rate. 5-Fluorouracil (5-FU) is still a widely used chemotherapeutic drug for advanced CRC, although the use of 5-FU is limited by drug resistance.20) In recent years, bevacizumab combined with different chemotherapeutics has been used to treat advanced CRC to improve overall survival, although the therapeutic effect was unsatisfactory.21) With the advent of immunotherapy, immune checkpoint inhibitors (ICIs) have been used to treat CRC. However, the efficacy of ICIs is limited to the microsatellite instability-high (MSI-H) subgroup, which accounts for 5% of advanced CRC. Therefore, new drugs are urgently required to explore CRC treatment.
CETP transfers cholesteryl esters between HDL and VLDL, IDL, and LDL to maintain cholesterol homeostasis. CETP is highly expressed in many cancers, such as colorectal, gastric, breast, pituitary adenoma and gallbladder cancers.8,22–25) Recent studies on the significance of CETP in breast cancer development have confirmed that CETP can help improve the survival outcomes of breast cancer cells as well as promote resistance to apoptosis. Furthermore, Mihajlovic et al. indicated that CETP activity increased during tumor progression in CRC. Therefore, inhibiting CETP is expected to be an effective treatment for CRC. In this study, we performed a series of experiments on CRC cells with the CETP inhibitor Evacetrapib to test this hypothesis. The results indicated that Evacetrapib suppressed CRC cell proliferation and induced cell apoptosis. This is the first study to indicate that Evacetrapib has antitumor effects on CRC cells.
Abnormally activated Wnt/β-catenin pathway is closely associated with CRC occurrence and development.26) Ren et al. reported that cholesterol activated the Wnt/β-catenin signaling pathway by binding with Dishevelled protein in tumors.27) Avasimibe, a drug that regulates cholesterol homeostasis, inhibits prostate cancer metastasis by inhibiting the Wnt/β-catenin pathway.28) However, whether the Wnt/β-catenin signaling pathway is involved in the role of Evacetrapib in CRC remains unclear. We found that Evacetrapib can inhibit the activity of Wnt luciferase, protein levels of p-β-catenin (Ser675), as well as mRNA expressions of downstream target genes of Wnt/β-catenin, including axin2, cyclinD1, c-myc and survivin. The above results indicate that inhibition of Wnt/β-catenin has a role in the effect of Evacetrapib in CRC.
The mitogen-activated protein kinase (MAPK) family includes three major subfamilies, extracellular signal-regulated kinase (ERK), JNK and p38 MAPK.29) JNK can be activated by cellular stresses and therefore plays an vital role in cell growth as well as apoptosis.30) For example, gambogic acid initiates apoptosis by activating the JNK signaling pathway.31) Apoptosis induced by costunolide in human leukemic cells depends on the activation of JNK signal.32) In this study, we further explored whether Evacetrapib-mediated apoptosis is associated with the JNK signaling pathway. The Western blot assay results showed that Evacetrapib activated the JNK pathway in CRC cells. In addition, the JNK inhibitor SP600125 significantly reversed the Evacetrapib-induced CRC cell apoptosis, implying a pro-apoptotic role of JNK activation by Evacetrapib in CRC cells. Conversely, Sancho et al. revealed that increased JNK activity accelerated tumorigenesis in CRC.33) Therefore, JNK might play a proapoptotic role based on specific stimuli.
The present study demonstrated that Evacetrapib plays an anti-colorectal cancer role by regulating the Wnt/β-catenin and JNK signaling pathways. Therefore, Evacetrapib is a potential candidate for CRC therapy. It is worth noting that Evacetrapib is a benzazepine that selectively and effectively inhibits the activity of CETP.34) Schmees et al. pointed out that benzazepine BAY6356 can inhibit the proliferation of acute myeloid leukemia and multiple myeloma cell lines.35) In this study, we did not detect the expression level of CETP in CRC cells and the change of CETP expression level after Evacetrapib treatment. Therefore, it is unclear whether the anti-CRC effect of Evacetrapib depends on CETP or benzazepine, which is worthy of further study. Further, silencing CETP can reduce the expression of intracellular cholesterol.36) It has been reported that cancer cells need high cholesterol levels for membrane biogenesis and other functional needs, suggesting that Evacetrapib may participate in the growth inhibition of CRC by reducing intracellular cholesterol.28) The present study did not test the cholesterol levels after treatment with Evacetrapib. However, our findings show a piece of previously unrecognized evidence for the role of Evacetrapib in CRC cells, which opens up new opportunities for treating CRC. Thus, it is necessary to further study the contributions of Evacetrapib in CRC.
Acknowledgments
This work was supported by Project to Attract High Level Foreign Experts (G20200019016); China Postdoctoral Scientific Foundation (2020M683114); Dongguan Science and Technology of Social Development Program (202050715001215); Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (20211405).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin., 71, 209–249 (2021).
- 2) Chu LY, Guo DM, Chen JT, Fang WK, Xie JJ, Peng YH, Xu YW, Li XX. The diagnostic value of serum L1CAM in patients with colorectal cancer. Technol. Cancer Res. Treat., 19, 1533033820920971 (2020).
- 3) Bardakhchyan S, Mkhitaryan S, Zohrabyan D, Safaryan L, Avagyan A, Harutyunyan L, Arakelyan J, Tamamyan G, Tananyan A. Treatment and outcomes of colorectal cancer in Armenia: a real-world experience from a developing country. JCO Glob. Oncol., 6, 1286–1297 (2020).
- 4) Latypova DK, Shmakov SV, Pechkovskaya SA, Filatov AS, Stepakov AV, Knyazev NA, Boitsov VM. Identification of spiro-fused pyrrolo[3,4-a]pyrrolizines and tryptanthrines as potential antitumor agents: synthesis and in vitro evaluation. Int. J. Mol. Sci., 22, 11997 (2021).
- 5) Ragusa S, Prat-Luri B, González-Loyola A, Nassiri S, Squadrito ML, Guichard A, Cavin S, Gjorevski N, Barras D, Marra G, Lutolf MP, Perentes J, Corse E, Bianchi R, Wetterwald L, Kim J, Oliver G, Delorenzi M, De Palma M, Petrova TV. Antiangiogenic immunotherapy suppresses desmoplastic and chemoresistant intestinal tumors in mice. J. Clin. Invest., 130, 1199–1216 (2020).
- 6) Huyghe N, Baldin P, Van den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol. Rep. (Oxf.), 8, 11–24 (2020).
- 7) Kühnast S, van der Tuin SJ, van der Hoorn JW, van Klinken JB, Simic B, Pieterman E, Havekes LM, Landmesser U, Lüscher TF, Willems van Dijk K, Rensen PC, Jukema JW, Princen HM. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J., 36, 39–48 (2015).
- 8) Zeljkovic A, Vekic J, Mihajlovic M, Gojkovic T, Vladimirov S, Zeljkovic D, Spasojevic-Kalimanovska V, Trifunovic B. Revealing the role of high-density lipoprotein in colorectal cancer. Int. J. Mol. Sci., 22, 3352 (2021).
- 9) Mihajlovic M, Gojkovic T, Vladimirov S, Miljkovic M, Stefanovic A, Vekic J, Zeljkovic D, Trifunovic B, Kotur-Stevuljevic J, Spasojevic-Kalimanovska V, Zeljkovic A. Changes in lecithin: cholesterol acyltransferase, cholesteryl ester transfer protein and paraoxonase-1 activities in patients with colorectal cancer. Clin. Biochem., 63, 32–38 (2019).
- 10) Lincoff AM, Nicholls SJ, Riesmeyer JS, et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med., 376, 1933–1942 (2017).
- 11) Lammers T, Peschke P, Ehemann V, Debus J, Slobodin B, Lavi S, Huber P. Role of PP2Calpha in cell growth, in radio- and chemosensitivity, and in tumorigenicity. Mol. Cancer, 6, 65 (2007).
- 12) Lee CH, Lee FY, Tarafder S, Kao K, Jun Y, Yang G, Mao JJ. Harnessing endogenous stem/progenitor cells for tendon regeneration. J. Clin. Invest., 125, 2690–2701 (2015).
- 13) Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, De Filippis L, Mangiola A, Maira G, Anile C, De Bonis P, Reynolds BA, Pasquale EB, Vescovi AL. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell, 22, 765–780 (2012).
- 14) Botchkina GI, Zuniga ES, Das M, Wang Y, Wang H, Zhu S, Savitt AG, Rowehl RA, Leyfman Y, Ju J, Shroyer K, Ojima I. New-generation taxoid SB-T-1214 inhibits stem cell-related gene expression in 3D cancer spheroids induced by purified colon tumor-initiating cells. Mol. Cancer, 9, 192 (2010).
- 15) Georgakilas AG, Martin OA, Bonner WM. p21: a two-faced genome guardian. Trends Mol. Med., 23, 310–319 (2017).
- 16) Li Y, Zhou Y, Wang M, Lin X, Zhang Y, Laurent I, Zhong Y, Li J. Ampelopsin inhibits breast cancer cell growth through mitochondrial apoptosis pathway. Biol. Pharm. Bull., 44, 1738–1745 (2021).
- 17) Singh RK, Lokeshwar BL. Depletion of intrinsic expression of Interleukin-8 in prostate cancer cells causes cell cycle arrest, spontaneous apoptosis and increases the efficacy of chemotherapeutic drugs. Mol. Cancer, 8, 57 (2009).
- 18) Jiang L, Wang Y, Liu G, Liu H, Zhu F, Ji H, Li B. C-Phycocyanin exerts anti-cancer effects via the MAPK signaling pathway in MDA-MB-231 cells. Cancer Cell Int., 18, 12 (2018).
- 19) Alotaibi AG, Li JV, Gooderham NJ. Tumour necrosis factor-α (TNF-α) enhances dietary carcinogen-induced DNA damage in colorectal cancer epithelial cells through activation of JNK signaling pathway. Toxicology, 457, 152806 (2021).
- 20) Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther., 206, 107447 (2020).
- 21) Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther., 5, 22 (2020).
- 22) Wang W, Min K, Chen G, Zhang H, Deng J, Lv M, Cao Z, Zhou Y. Use of bioinformatic database analysis and specimen verification to identify novel biomarkers predicting gastric cancer metastasis. J. Cancer, 12, 5967–5976 (2021).
- 23) Esau L, Sagar S, Bangarusamy D, Kaur M. Identification of CETP as a molecular target for estrogen positive breast cancer cell death by cholesterol depleting agents. Genes Cancer, 7, 309–322 (2016).
- 24) Sidaraite A, Liutkeviciene R, Glebauskiene B, Vilkeviciute A, Kriauciuniene L. Associations of cholesteryl ester transfer protein (CETP) gene variants with pituitary adenoma. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub., 164, 189–195 (2020).
- 25) Zhang Y, Liu Y, Duan J, Wang H, Zhang Y, Qiao K, Wang J. Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle, 18, 3337–3350 (2019).
- 26) Zhang Y, Guo L, Li Y, Feng GH, Teng F, Li W, Zhou Q. MicroRNA-494 promotes cancer progression and targets adenomatous polyposis coli in colorectal cancer. Mol. Cancer, 17, 1 (2018).
- 27) Sheng R, Kim H, Lee H, Xin Y, Chen Y, Tian W, Cui Y, Choi JC, Doh J, Han JK, Cho W. Cholesterol selectively activates canonical Wnt signalling over non-canonical Wnt signalling. Nat. Commun., 5, 4393 (2014).
- 28) Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat. Metab., 2, 132–141 (2020).
- 29) Guldal CG, Ahmad A, Korshunov A, Squatrito M, Awan A, Mainwaring LA, Bhatia B, Parathath SR, Nahle Z, Pfister S, Kenney AM. An essential role for p38 MAPK in cerebellar granule neuron precursor proliferation. Acta Neuropathol., 123, 573–586 (2012).
- 30) Lin Y, Peng N, Li J, Zhuang H, Hua ZC. Herbal compound triptolide synergistically enhanced antitumor activity of amino-terminal fragment of urokinase. Mol. Cancer, 12, 54 (2013).
- 31) Wen C, Huang L, Chen J, Lin M, Li W, Lu B, Rutnam ZJ, Iwamoto A, Wang Z, Yang X, Liu H. Gambogic acid inhibits growth, induces apoptosis, and overcomes drug resistance in human colorectal cancer cells. Int. J. Oncol., 47, 1663–1671 (2015).
- 32) Choi JH, Lee KT. Costunolide-induced apoptosis in human leukemia cells: involvement of c-jun N-terminal kinase activation. Biol. Pharm. Bull., 32, 1803–1808 (2009).
- 33) Sancho R, Nateri AS, de Vinuesa AG, Aguilera C, Nye E, Spencer-Dene B, Behrens A. JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J., 28, 1843–1854 (2009).
- 34) Shrestha S, Wu BJ, Guiney L, Barter PJ, Rye KA. Cholesteryl ester transfer protein and its inhibitors. J. Lipid Res., 59, 772–783 (2018).
- 35) Schmees N, Haendler B, Lejeune P, Stresemann A, Neuhaus R, Siegel S, Fernandez-Montalvan AE, Weinmann H, Gekeler V. Abstract 4749. New benzazepine BET-inhibitors with improved oral bioavailability. Cancer Res., 74, 4749–4749 (2014).
- 36) Izem L, Morton RE. Possible role for intracellular cholesteryl ester transfer protein in adipocyte lipid metabolism and storage. J. Biol. Chem., 282, 21856–21865 (2007).