Abstract
Vascular endothelial growth factor (VEGF) is a key mediator of angiogenesis, which plays a key role in the proliferation, migration and invasion of endothelial cell. Bisdemethoxycurcumin (BDMC) is a natural demethoxy curcumin derivative. In this study, we explored the mechanisms whereby BDMC is able to influence the proliferative, migratory and invasive activity of human umbilical vein endothelial cells (HUVECs) in response to VEGF treatment. These experiments revealed that BDMC at 10 and 20 µM suppressed HUVECs proliferation in response to VEGF (10 ng/mL) without impacting the proliferation in absence of VEGF. BDMC treatment also signifantly suppressed VEGF-induced migratory and invasive activity in HUVECs. However, the selective AMP-activated protein kinase (AMPK) inhibitor compound C (3 µM) treatment signifantly reversed all of these effects. Flow cytometric assay showed BDMC treatment was found to induce G0/G1 phase cell cycle arrest. Western blotting further indicated that BDMC treatment increased the ratios of p-AMPK/AMPK and LC3B/LC3A, up-regulated the expression of Beclin-1, decreased the ratio of p-mammalian target of rapamycin (mTOR)/mTOR, down-regulated the expression of cyclin D1 and CDK4. Overall, these data suggested that BDMC may exert benefical effect on HUVECs activation by activating autophagy and inducing cell cycle arrest through regulation of the AMPK/mTOR pathway, which could provide a potential compound candidate for the treatment of diseases related to VEGF overproduction.
INTRODUCTION
The endothelial cell layer is integral to the regulation of vascular homeostasis, controlling parameters including the proliferation of smooth muscle cells, coagulation, and arterial tone.1) It is reported that increased production of vascular endothelial growth factor (VEGF) palys a key role in vascular endothelial dysfunction, which is closely linked to conditions including rheumatoid arthritis (RA), atherosclerosis, inflammatory bowel disease and cancer.2,3) Such as, several recent reports have shown the expression of VEGF to be dysregulated in RA, with high levels of this growth factor in synovial fluid suggesting that it may be a pathogenic factor in this autoimmune context.3) Functionally, VEGF can bind to endothelial receptors and thereby increase vascular permeability and plasma protein leakage into the joints.4,5)
The cell cycle and autophagic activity are highly regulated pathways that are critical to tissue development, homeostasis, and protection against disease.5) Autophagy is a process whereby cells degrade organelles and macromolecular structures to maintain homeostatic set-points in the context of stressful conditions, with this degradative activity being critical to cellular structural and functional integrity.6) In vitro, autophagy activiation and cell cycle arrest reportedly suppresses the proliferation of endothelial cells in response to atherogenic stimuli.7) Moreover, drug-eluting stents are frequently loaded with mammalian target of rapamycin (mTOR) inhibitor or related drugs capable of inducing autophagy and thus preventing endothelial proliferation.8) Engaging autophagic activity may therefore represent a viable approach to preventing or treating certain diseases, inhibiting lesion progression and stability.9) Once autophagy is triggered via the AMP-activated protein kinase (AMPK) pathways, LC3B protein levels rise to facilitate autophagosome formation.10)
Natural compounds have been reported to be of value in the treatment of many diseases, with curcuminoids in particular exhibiting antioxidant, antitumor, and anti-inflammatory properties with clinical promise.11) The curcumin derivative bisdemethoxycurcumin (BDMC) has been reported to exhibit improved obvious uptake and stability relative to unmodified curcumin, yet there have been few studies to date exploring its beneficial properties.12) Some research suggests that BDMC can suppress tumor cell proliferation and viability via the induction of cell cycle arrest, in addition to inhibiting the invasive and metastatic activity of these cells by inducing autophagy.13)
Despite these promising reports in oncological contexts, few research has focused on the mechanisms whether BDMC treatment may regulate vascular endothelial dysfunction. As such, the present study was formulated to explore the ability of BDMC to prevent VEGF-induced stimulation of proliferative, migratory and invasive activity in human umbilical vein endothelial cells (HUVECs) and to clarify the underlying molecular mechanisms.
MATERIALS AND METHODS
Cells and ReagentsHUVECs were purchased from American Type Culture Collection (ATCC) (Manassas, U.S.A.). Cells were grown in endothelial cell medium (Gibco, U.S.A.) containing 10% fetal bovine serum (Gibco, U.S.A.) and penicillin/streptomycin (100 U/mL) in a humidified 37 °C 5% CO2 incubator. Cells were treated with recombinant human VEGF for 24 h at a dose of 10 ng/mL.
BDMC (C19H16O4, MW = 308.33) was obtained from Tokyo Chemical Industry Co., LTD. (Tokyo, Japan). Compound C was from MedChemExpress Co., LTD. (Shanghai, China). Recombinant Human VEGF (VEGF-A) and Cell-Counting Kit 8 (CCK8) were from Solarbio Science & Technology Co., Ltd. (Beijing, China). Antibodies specific for phosphorylated AMPK (p-AMPK), total AMPK, phosphorylated mTOR (p-mTOR), total mTOR, LC3A/B, Beclin-1, cyclin D1, CDK4 and β-actin were from Abcam. Annexin V-fluorescein isothiocyanate (FITC)/PE Apoptosis kit, propidium iodide (PI) cell cycle kit, and matrigel were from BD Biosciences (CA, U.S.A.).
Proliferation AssayHUVECs were plated in 96-well plates (104/well) and allowed to adhere for 12 h. The next 24 h period, cells were treated with VEGF (10 ng/mL) or vehicle cell medium (control group), in the presence or absence of BDMC (1–20 µM) and compound C (3 µM). Then, each well was supplemented with 10 µL of CCK-8 reagent followed by an additional 2 h incubation at 37 °C. Absorbance was then measured at 450 nm using a Microplate Reader (Molecular Devices M3, U.S.A.).14)
Migration AssayHUVECs were grown to 80% confluence in 6-well plates that had been coated with 0.1% gelatin (Sigma-Aldrich, MO, U.S.A.). The next 24 h period, cells were treated with VEGF (10 ng/mL) or vehicle cell medium (control group), in the presence or absence of BDMC (10 or 20 µM) and compound C (3 µM). Then, 1 mL pipette tip was used to scratch the monolayer surface, cells were rinsed using phosphate buffered saline (PBS) and added serum-free medium. After 24 h, imaged using an inverted microscope (Olympus CX71).15)
Invasion AssayTranswell inserts (BD Biosciences) were used for invasion assays as in previous study.16) Briefly, HUVECs were added atop the matrigel-coated transwell filter, while media containing 10% fetal bovine serum was added to the lower chamber. cells were treated with VEGF (10 ng/mL) or vehicle cell medium (control group), in the presence or absence of BDMC (10 or 20 µM) and compound C (3 µM) for 24 h. A cotton swab was used to remove non-invasive cells from the upper chamber, with the remaining invasive cells being fixed, stained using crystal violet, and imaged (Olympus CX71).
Cell Apoptosis AssayAn Annexin V-FITC/PI apoptosis kit was used based on provided instructions to analyze cell apoptotisis and necrosis. Briefly, cells were rinsed in PBS and resuspended in 100 µL of binding buffer to which Annexin V-FITC (5 µL) was then added, followed by incubation for 5 min while protected from light. Cells were then incubated with 5 µL propidium iodide for an additional 15 min, after which a flow cytometer (BD FACSCanto II, U.S.A.) was used to analyze the stained cells.
Cell Cycle AssayCells were plated overnight in 6-well plates, treated with drugs in triplicate as above for 24 h, and then harvested and transferred into flow cytometry tubes. Cells were then fixed for 1 h in 70% ethanol at −20 °C, rinsed two times using cold PBS, and stained at 37 °C with PI solution (50 µg/mL PI in H2O, 0.1% Triton-X100, 0.1% trisodium citrate dehydrate, 6.25 µg/mL Ribonuclease A (RNase A) for 40 min. Cells were then analyzed via flow cytometry (BD FACSCanto II, U.S.A.) within 3 h.
Immunofluorescence AnalysisHUVECs were incubated with anti-p-AMPK antibody (1 : 500), followed by incubation with the corresponding secondary antibodies overnight. Then Cells were counterstained with 4′-6-diamidino-2-phenylindole (DAPI) to visualize the nucleus (blue). Finally, cells were evaluated by Zeiss (LSM800, Germany) confocal microscope.
Western BlottingAfter treatment for 24 h with indicated doses of BDMC and compound C, cells were rinsed using PBS and lysed for 30 min with a lysis buffer (0.5 M NaCl, 50 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.05% sodium dodecyl sulfate (SDS), 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), pH 7.4) on ice. Lysates were then centrifuged for 10 min at 12000 × g at 4 °C, with a bicinchoninic acid (BCA) Protein Assay Kit (Beyotime, China) being used to measure protein concentrations in collected supernatants. Proteins were separated via 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes that were blocked using 5% non-fat milk, followed by incubation overnight with appropriate antibodies at 4 °C. Following a washing step, blots were probed with HRP-conjugated goat anti-rabbit secondary antibodies for 1 h, washed thrice with Tris-buffered saline (TBS)-T, and imaged with an enhanced chemiluminescence reagent before detection with a gel imaging platform (GE, LAS4000). ImageQuant LAS4000 V1.22 was used for semi-quantitative densitometric analyses.
Statistical AnalysisData are means ± standard deviation (S.D.) and were compared via one-way ANOVAs with Tukey’s multiple comparison test. p < 0.05 was the significance threshold, and all experiments were repeated three times.
RESULTS
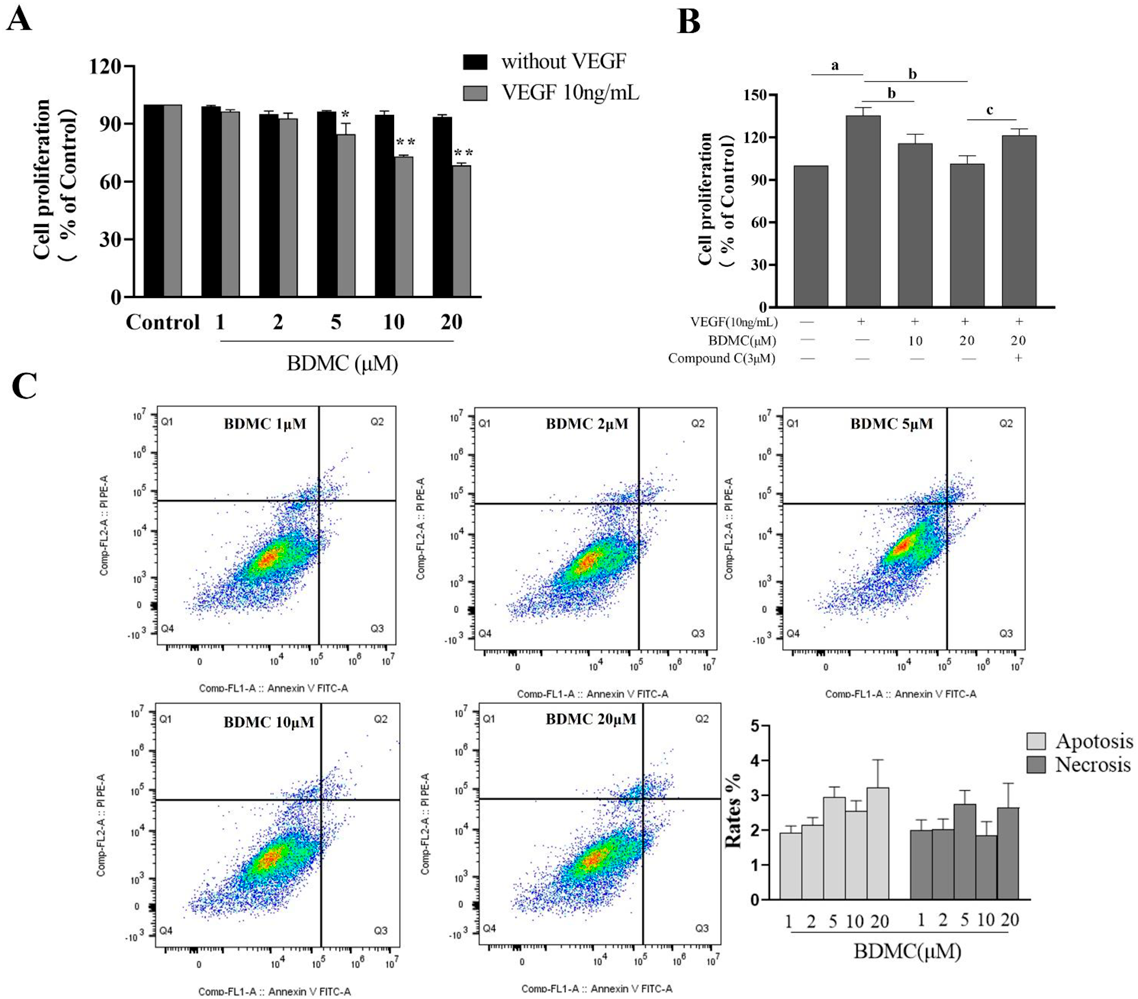
BDMC Suppresses HUVECs Proliferation in Response to VEGF Treatment, Rather than Induces ApoptosisAs shown in Fig. 1A, BDMC at 5, 10, and 20 µM significantly suppressed the proliferation of HUVECs following treatment with VEGF for 24 h in a dose-dependent manner (p < 0.05, p < 0.01), without affecting basal HUVECs proliferation in absence of VEGF (all p > 0.05). As shown in Fig. 1B, compared to vehile control group, the treatment of cultured HUVECs for 24 h treated with VEGF resulted in significantly increased proliferation (p < 0.01), whereas BDMC 10 and 20 µM groups showed a dose-dependent decrease in the proferation (p < 0.01). However, compound C was able to reverse the antiproliferative effect of BDMC treatment (p < 0.01). As shown in Fig. 1C, flow cytometry shows that the rates of apoptosis and necrosis of HUVECs in BDMC (1–20 µM) treated groups were no significant differences between each group (all p > 0.05).
BDMC Inhibits VEGF-Induced HUVECs MigrationAs shown in Fig. 2, VEGF treatment of HUVECs promotes cell migration compared to control group (p < 0.01). Compared to VEGF treatment group, BDMC at 10 and 20 µM inhibited the migration of HUVECs in a dose-dependent manner (p < 0.01), while compound C at the dose of 3 µM reversed the effect of BDMC (p < 0.01).
BDMC Suppresses VEGF-Induced HUVECs InvasionAs shown in Fig. 3, VEGF treatment of HUVECs promotes cell invasion compared to control group (p < 0.01). Compared to VEGF treatment group, BDMC at 10 and 20 µM inhibited the invasion of HUVECs in a dose-dependent manner (p < 0.01), while compound C at the dose of 3 µM reversed the effect of BDMC (p < 0.01).
BMDC Promotes Cell Cycle Arrest in VEGF-Treated HUVECsAs shown in Fig. 4A, an analysis of cell cycle progression revealed that the ratio of cells in the G0/G1 phase in HUVECs treated with VEGF showed a significant decrease compared to control group (p < 0.01). BDMC-mediated inhibition of HUVECs proliferative activity was tied to a concentration-dependent increase in G0/G1 phase arrest, with BDMC doses of both 10 and 20 µM significantly increasing the ratio of cells in the G0/G1 phase (p < 0.01), which support a role for the induction of cell cycle arrest as a mediator of the antiproliferative activity of BDMC. However, compound C was sufficient to significantly reverse these BDMC-induced effects (p < 0.01).
As shown in Fig. 4B, the expression of cyclin D1 and CDK4 in HUVECs treated with VEGF showed a significant increase compared to control group (p < 0.01). Compared to cells in the VEGF group, HUVECs treated with BDMC exhibited significant decrease in the expression of cyclin D1 and CDK4 (p < 0.05, p < 0.01). Compared to cells in the BDMC 20 µM group, there was a significant increase in the expression of cyclin D1 and CDK4 in the BDMC 20 µM + compound C 3 µM treatment group (p < 0.01).
BMDC Activates AMPK/mTOR Pathway and Regulates the Expression of Autophagy-Associated Proteins in VEGF-Treated HUVECsAs shown in Fig. 5A, the fluorescence intensity of p-AMPK in BDMC 20 µM treatment group was higher compared to VEGF group, whereas compound C treatment reversed the effect of BDMC. As shown in Fig. 5B, exposure of HUVECs to VEGF resulted in suppression of AMPK and enhancement of mTOR as demonstrated by the significant decrease in the ratio of p-AMPK/AMPK and increase in the ratio of p-mTOR/mTOR (p < 0.01). Compared to VEGF group, HUVECs treated with BDMC exhibited significant increase in the ratio of p-AMPK/AMPK and decrease in the ratio of p-mTOR/mTOR (p < 0.01), both of these ratios were significantly reversed in BDMC 20 µM + compound C 3 µM treatment group (p < 0.01). Analyses of autophagy-related protein expression revealed that there was a significant decrease in the ratio of LC3B/LC3A and expression of Beclin-1 in HUVECs treated with VEGF compared to control group (p < 0.01). BDMC treatment significantly increased the ratio of LC3B/LC3A (p < 0.05, p < 0.01) and the expression of Beclin-1 (p < 0.01), whereas compound C treatment reversed these effects (p < 0.05, p < 0.01).
DISCUSSION
Endothelial dysfunction has been identified as a key pathogenic driver of conditions including RA, atherosclerosis and cancer.17) Natural compounds represent an invaluable source of pharmacological products for use in drug discovery efforts owing to their high degree of structural diversity and functional bioactivity.18,19) BDMC is a natural curcumin derivative that exhibits antiproliferative and anti-inflammatory properties. The results of the present study provides novel evidence that BDMC can suppress HUVECs proliferation, invasion, and migration induced in response to VEGF treatment in a dose-dependent fashion. In contrast, the AMPK inhibitor compound C was able to reverse these BMDC treatment-related effects.
We found that treatment with BDMC was sufficient to inhibit VEGF-incubated HUVECs proliferation in a dose-dependent manner without impacting the basal proliferation of cells, consistent with the specific antiproliferative blockade of VEGF-induced stimulation. Meanwhile, we also observed dose-dependent reductions in the migratory and invasive activity of HUVECs with VEGF upon BDMC treatment. These results demonstrated BDMC is able to reduce the main biological actions (endothelial cell proliferation, migration and invasion) induced by VEGF, which indicated that BDMC may exert its effect through directly acting on targets activated/supprested by VEGF.
Autophagy is an important regulatory pathway in a range of stress-related contexts, regulating the survival or death of individual cells, with clear evidence suggesting the importance of this stress response mechanism in the endothelium.20) Autophagy can contribute to the reversible inhibition of cellular proliferation for both vascular smooth muscle cells and other cell lines.21) During autophagy, LC3A in the cytoplasm is converted to LC3B, which is in the inner and outer membranes of autophagosomes, so that the ratio of LC3B/LC3A could also be used to gauge the degree of autophagic activity.21–23) Here, we found the ratio of LC3B/LC3A in significantly increased in BDMC treated groups, that is to say BDMC could enhance the autophagy level in HUVEC treated with VEGF. Beclin-1 is an important mediator of autophagosome development and maturation in this process, and we found that BDMC promoted increases in the expression of Beclin-1 further supporting autophagosome formation relative to that observed in VEGF-treated cells. AMPK phosphorylation has been reported to inhibit signaling via the mTOR pathway, thus regulating cellular migration, differentiation, and proliferation.24) AMPK/mTOR signaling plays an important role in the process of autophagy.25) We therefore explored the activation of the AMPK/mTOR ratio in order to better understand the mechanisms whereby BDMC induces HUVECs autophagy, revealing an increase in the p-AMPK/AMPK ratio and a decrease in the p-mTOR/mTOR in HUVECs following BDMC treatment, Importantly, compound C, which inhibits AMPK, reversed the observed BDMC-induced autophagic activity and increased the p-mTOR/mTOR expression in HUVECs, in addition to reversing observed changes in Beclin-1 expression and LC3 conversion.
The cell cycle is a fundamental process that is tightly regulated to ensure cellular and tissue homeostasis.26) AMPK-induced mTOR activity can alter the activation and expression of cyclins, cyclin-dependent kinases, and inhibitors thereof.27) The activation of AMPK has been shown to induce cell cycle arrest in a range of cell types including SKM-1, HepG2, and murine embryonic fibroblast cells.28) The cyclins D1 bind to and activate CDK4 preferentially, which plays a critical role in G0/G1 phase progression.27) BDMC down-regulated the expression of cyclin D1 and CDK4 in VEGF-treated HUVECs. Also, increased expression of cyclin D1 and CDK4 was diminished by compound C treatment. Because the expression of cyclin D1 and CDK4 is associated with activation of AMPK/mTOR, our results suggest that BDMC inhibits VEGF-treated HUVECs cycle progression at G0/G1 phase by through activation of AMPK/mTOR pathway inducing down-regulation of cyclin D1 and CDK4. However, further research will be necessary to elucidate the interplay between AMPK/mTOR signaling and the regulation of cyclin-related factors in BDMC-treated HUVECs.
Therapeutic approaches simultaneously targeting the cell cycle and autophagy have recently been highlighted as promising strategies.29) We found that BDMC treatment (10 and 20 µM) was sufficient to induce endothelial cell autophagic activity while promoting G0/G1 phase cell cycle arrest. There are many cross-regulatory interactions between the autophagic and cell cycle pathways, further research will be essential to fully clarify the mechanisms whereby BDMC is able to simultaneously engage both of these pathways to exert its therapeutic efficacy.
The present data suggest that BDMC can efficiently inhibit the activity of VEGF in vitro, counteracting inducible endothelial cell proliferation, invasion, and migration and thus providing a theoretical foundation explaining the therapeutic benefits associated with this compound. Importantly, the dual-inhibitory mechanism of action whereby BDMC suppresses VEGF-induced activity highlights its promise as a therapeutic agent for treating a range of conditions in which pathological VEGF overproduction is observed, including atherosclerosis, diabetic retinopathy, cancer and so on.
Overall, these results suggested that BDMC could inhibit the proliferative, migratory, and invasive activity of HUVECs in response to VEGF treatment by inducing cell cycle arrest and autophagic activity via AMPK/mTOR pathway in vitro. These results offer novel insight into the antiproliferative effects of such BDMC treatment, highlighting its potential clinical utility. However, further research will be essential to better define how to induce beneficial intermediate levels of cell cycle arrest and autophagy in vivo.
Acknowledgments
This study was supported by Natural Science Foundation of Shandong Province (No. ZR2017PH073). The authors would like to thank all the reviewers who participated in the study.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Gimbrone MA Jr, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol., 22, 9–15 (2013).
- 2) Liu W, Ou Y, Yang Y, Zhang X, Huang L, Wang X, Wu B, Huang M. Inhibitory effect of punicalagin on inflammatory and angiogenic activation of human umbilical vein endothelial cells. Front. Pharmacol., 12, 727920 (2021).
- 3) Wang L, Wang S, Xue A, Shi J, Zheng C, Huang Y. Thalidomide inhibits angiogenesis via downregulation of VEGF and angiopoietin-2 in crohn’s disease. Inflammation, 44, 795–807 (2021).
- 4) Schwager S, Detmar M. Inflammation and lymphatic function. Front. Immunol., 10, 308 (2019).
- 5) Han YH, Mun JG, Jeon HD, Kee JY, Hong SH. Betulin inhibits lung metastasis by inducing cell cycle arrest, autophagy, and apoptosis of metastatic colorectal cancer cells. Nutrients, 12, 66 (2019).
- 6) Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell, 177, 1682–1699 (2019).
- 7) Schaaf MB, Houbaert D, Meçe O, Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death Differ., 26, 665–679 (2019).
- 8) Djajadikerta A, Keshri S, Pavel M, Prestil R, Ryan L, Rubinsztein DC. Autophagy induction as a therapeutic strategy for neurodegenerative diseases. J. Mol. Biol., 432, 2799–2821 (2020).
- 9) Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol., 17, 802–815 (2018).
- 10) Runwal G, Stamatakou E, Siddiqi FH, Puri C, Zhu Y, Rubinsztein DC. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep., 9, 10147 (2019).
- 11) Guntuku L, Naidu VG, Yerra VG. Mitochondrial dysfunction in gliomas: pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol., 14, 567–583 (2016).
- 12) Jäger R, Lowery RP, Calvanese AV, Joy JM, Purpura M, Wilson JM. Comparative absorption of curcumin formulations. Nutr. J., 13, 11 (2014).
- 13) Chen J, Li L, Su J, Chen T. Natural borneol enhances bisdemethoxycurcumin-induced cell cycle arrest in the G2/M phase through up-regulation of intracellular ROS in HepG2 cells. Food. Funct., 6, 740–748 (2015).
- 14) Chen J, Chen T, Zhu Y, Li Y, Zhang Y, Wang Y, Li X, Xie X, Wang J, Huang M, Sun X, Ke Y. circPTN sponges miR-145-5p/miR-330-5p to promote proliferation and stemness in glioma. J. Exp. Clin. Cancer Res., 38, 398 (2019).
- 15) Hossian AKMN, Mattheolabakis G. Cellular migration assay: an in vitro technique to simulate the wound repair mechanism. Methods Mol. Biol., 2193, 77–83 (2021).
- 16) Pijuan J, Barceló C, Moreno DF, Maiques O, Sisó P, Marti RM, Macià A, Panosa A. In vitro cell migration, invasion, and adhesion assays: from cell imaging to data analysis. Front. Cell Dev. Biol, 7, 107 (2019).
- 17) Konukoglu D, Uzun H. Endothelial dysfunction and hypertension. Adv. Exp. Med. Biol., Vol. 956, Springer, New York, pp. 511–540 (2017).
- 18) Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov., 14, 111–129 (2015).
- 19) Rodrigues T, Reker D, Schneider P, Schneider G. Counting on natural products for drug design. Nat. Chem., 8, 531–541 (2016).
- 20) Cui B, Lin H, Yu J, Yu J, Hu Z. Autophagy and the immune response. Adv. Exp. Med. Biol., 1206, 595–634 (2019).
- 21) Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol., 66, 89–100 (2020).
- 22) Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature, 469, 323–335 (2011).
- 23) Xu HD, Qin ZH. Beclin 1, Bcl-2 and autophagy. Adv. Exp. Med. Biol., 1206, 109–126 (2019).
- 24) Zhang JJ, Li YQ, Wang YS, Chen L, Wang XZ. Estradiol ameliorates metformin-inhibited Sertoli cell proliferation via AMPK/TSC2/mTOR signaling pathway. Theriogenology, 175, 7–22 (2021).
- 25) Li MY, Zhu XL, Zhao BX, Shi L, Wang W, Hu W, Qin SL, Chen BH, Zhou PH, Qiu B, Gao Y, Liu BL. Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis., 10, 489 (2019).
- 26) Deng X, Sheng JX, Liu H, Wang NN, Dai CJ, Wang ZG, Zhang J, Zhao JY, Dai EQ. Cinobufagin promotes cell cycle arrest and apoptosis to block human esophageal squamous cell carcinoma cells growth via the p73 signalling pathway. Biol. Pharm. Bull., 42, 1500–1509 (2019).
- 27) Wang X, Lin Y, Kemper T, Chen J, Yuan Z, Liu S, Zhu Y, Broering R, Lu M. AMPK and Akt/mTOR signalling pathways participate in glucose-mediated regulation of hepatitis B virus replication and cellular autophagy. Cell. Microbiol., 22, e13131 (2020).
- 28) Wang ST, Ho HJ, Lin JT, Shieh JJ, Wu CY. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis., 8, e2626 (2017).
- 29) Li C, Imai M, Hasegawa S, Yamasaki M, Takahashi N. Growth inhibition of refractory human gallbladder cancer cells by retinol, and its mechanism of action. Biol. Pharm. Bull., 40, 495–503 (2017).