Abstract
An increase in intracellular Ca2+ concentration ([Ca2+]i) activates Ca2+-sensitive enzymes such as Ca2+/calmodulin-dependent kinases (CaMK) and induces gene transcription in various types of cells. This signaling pathway is called excitation-transcription (E-T) coupling. Recently, we have revealed that a L-type Ca2+ channel/CaMK kinase (CaMKK) 2/CaMK1α complex located within caveolae in vascular smooth muscle cells (SMCs) can convert [Ca2+]i changes to gene transcription profiles that are related to chemotaxis. Although CaMK1α is expected to be the key molecular identity that can transport Ca2+ signals originated within caveolae to the nucleus, data sets directly proving this scheme are lacking. In this study, multicolor fluorescence imaging methods were utilized to address this question. Live cell imaging using mouse primary aortic SMCs revealed that CaMK1α can translocate from the cytosol to the nucleus; and that this movement was blocked by nifedipine or a CaMKK inhibitor, STO609. Experiments using two types of Ca2+ chelators, ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), combined with caveolin-1 knockout (cav1-KO) mice showed that local Ca2+ events within caveolae are required to trigger this CaMK1α nuclear translocation. Importantly, overexpression of cav1 in isolated cav1-KO myocytes recovered the CaMK1α translocation. In SMCs freshly isolated from mesenteric arteries, CaMK1α was localized mainly within caveolae in the resting state. Membrane depolarization induced both nuclear translocation and phosphorylation of CaMK1α. These responses were inhibited by nifedipine, STO609, cav1-KO, or BAPTA. These new findings strongly suggest that CaMK1α can transduce Ca2+ signaling generated within or very near caveolae to the nucleus and thus, promote E-T coupling.
INTRODUCTION
Changes in intracellular Ca2+ concentration ([Ca2+]i) contribute to many essential biological responses such as neurotransmission, hormone secretion, and muscle contraction.1,2) An increase in [Ca2+]i activates Ca2+-dependent enzymes, then resulting in activation of transcription factors and subsequent changes in patterns of gene transcription.3) This signaling pathway is referred to as excitation-transcription (E-T) coupling. It has been reported that E-T coupling is one of the mechanisms responsible for late phase long-term synaptic plasticity and memory in neurons.3) However, E-T coupling is also involved in pathological processes such as myocardial remodeling in heart.4) In smooth muscle tissues, E-T coupling directly associated with Ca2+ influx through Cav1.2, a dominant L-type voltage dependent Ca2+ channel isoform in vascular smooth muscle cells (SMCs), has been reported to play important roles in physiological5) or pathophysiological6,7) functions, such as differentiation and hypertension.
Directional Ca2+ signaling from the plasma membrane to the nucleus is an important step during E-T coupling pathways. Published data have revealed that several Ca2+-dependent kinases, i.e., Ca2+/CaM-dependent kinases (CaMK), can contribute to this signal transduction by phosphorylating downstream molecules8–10) or by shuttling Ca2+/CaM into the nucleus.11,12) The CaMK1 family is composed of four members, which are coded by four different genes: CAMK1, PNCK, CAMK1G, and CAMK1D, which produce CaMK1α, CaMK1β, CaMK1γ, and CaMK1δ proteins, respectively. Binding of Ca2+/CaM to CaMK1 exposes an “activation loop” site to allow phosphorylation by the upstream CaMK kinase (CaMKK) when simultaneously activated by Ca2+/CaM.13) Phosphorylation of the activation loop in CaMK1 primarily increases their Ca2+/CaM-dependent enzymatic activities. In neurons, this CaMKK-mediated CaMK1 phosphorylation can persist for up to an hour or more.14) However, CaMK1α lacks autonomous activity by auto-phosphorylation. Therefore, kinase activity of phosphorylated CaMK1α (pCaMK1α) decreases as [Ca2+]i returns to basal levels, although a small amount of activation remains as long as Thr177 is phosphorylated.15) CaMK1α is mainly localized in cytosol because of its nuclear export signal (NES) at C terminus.16) CaMK1α is known to be activated after Ca2+ influx through N-methyl-D-aspartate (NMDA) receptors in hippocampal neurons and results in activity-dependent spine formation/maturation9) and dendritic outgrowth and branching.17) CaMK1α also phosphorylates microtubule affinity regulating kinase-2 to regulate neurite outgrowth in hippocampus.18) Interestingly, the activation of γ-aminobutyric acid (GABA) receptor selectively stimulates elongation of axons by driving CaMKK-CaMK1α cascade, while brain derived neurotrophic factor (BDNF) shows selective growth of dendrite through CaMKK-CaMK1γ pathway.19) In smooth muscle tissues, however, the role of CaMK1α is not fully understood.
Recently, we have demonstrated that a molecular complex consisting of Cav1.2/CaMKK2/CaMK1α located within caveolae, can induce cAMP response element binding protein (CREB)-dependent gene expression that is related to chemotaxis and inflammation in vascular smooth muscles. This results in macrophage accumulation and medial hypertrophy in vessels after sustained pressure loading.20) In this previous study, CaMK1α was suggested to be the prime candidate to deliver Ca2+ signals generated at the cell surface to the nucleus; however, direct evidence was not presented. In addition, it was unclear if CaMK1α actually translocates from the cell surface/caveolae to the nucleus in freshly-isolated vascular SMCs. Data obtained in the present study establishes that CaMK1α can translocate from the cytosol to the nucleus in response to membrane depolarization and subsequent [Ca2+]i elevation in mouse primary aortic SMCs (ASMCs). This nuclear translocation and phosphorylation of CaMK1α requires Ca2+ influx through Cav1.2 channels and the activation of CaMKK2 within caveolae in both primary ASMCs and freshly-isolated mesenteric arterial SMCs (MASMCs). These new findings strongly suggest that CaMK1α transports Ca2+ signals that originate at specific sites (caveolae) to the nucleus to initiate E-T coupling in vascular SMCs.
MATERIALS AND METHODS
AnimalsWild type (WT) and caveolin1-knockout (cav1-KO) mice (C57BL/6 background, 8-15 weeks, male or female) were obtained from Japan SLC (Hamamatsu, Japan) and Jackson Labs (Bar Harbor, ME, U.S.A.), respectively.20) All experiments were approved by the Ethics Committee of Nagoya City University (H30-P-1) and conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Japanese Pharmacological Society.
Single Cell Isolation and Unpassaged SMC CultureTo obtain unpassaged primary ASMCs,20) SMCs were isolated from mouse aorta. These isolated myocytes were then seeded on glass bottom dishes (Matsunami, Osaka, Japan) coated with 20 µg/mL laminin (MilliporeSigma, St. Louis, MO, U.S.A.) for 2 h and kept in an incubator at 37 °C with 5% CO2 for 5–7 d to allow expansion to a semi-confluent state. Then, the medium was switched to Dulbecco’s modified Eagle’s medium (DMEM) (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) with 0.1% fetal bovine serum (FBS) (MilliporeSigma), 1 × ITS (insulin, transferrin and selenium, FUJIFILM Wako Pure Chemical Corporation) supplement and 200 µM L-ascorbic acid sodium (FUJIFILM Wako Pure Chemical Corporation) for an additional 2–3 d. Adenovirus vectors (ViraPower™Adenoviral Expression System, Thermo Fisher Scientific, Waltham, MA, U.S.A.)20) were applied to the myocytes when the medium was switched. Single MASMCs were freshly isolated from second- and third-order mesenteric arteries as reported previously.20) These isolated cells were incubated with standard (5 mM KCl) N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)-buffered solution: (mM) 137 NaCl, 5.9 KCl, 2.2 CaCl2, 1.2 MgCl2,14 glucose, and 10 HEPES or 60 mM KCl HEPES-buffered solution: (mM) 83 NaCl, 60 KCl, 2.2 CaCl2, 1.2 MgCl2,14 glucose, and 10 HEPES in the presence of vehicle or drugs. The pH was adjusted to 7.4 with NaOH.
Transfection Using Adenovirus VectorsCaMK1α and cav1 were labeled with green fluorescent protein (GFP) and mCherry, respectively at the N termini. mCherry was used as a mock control. These constructs were cloned into pENTR1A and obtained pAd/CMV/V5-DEST vectors using Gateway system (Thermo Fisher Scientific).20) Adenovirus vectors were harvested from 293 A cells (Thermo Fisher Scientific) after introducing linearized pAd/CMV/V5-DEST. Primary ASMCs at semiconfluent condition were infected for 12 h and then washed. Experiments were performed 48–72 h after infection.
ImmunocytochemistryFreshly-isolated MASMCs were fixed with 4% paraformaldehyde (PFA) (FUJIFILM Wako Pure Chemical Corporation) in phosphate-buffered saline as reported previously.21) These were treated with 0.2% Triton X-100 (FUJIFILM Wako Pure Chemical Corporation), 3% bovine serum albumin (BSA, MilliporeSigma), and anti-CaMK1 rabbit monoclonal antibody (1 : 300, HPA051409, MilliporeSigma), anti-CaMKI (phosphor-Thr177) rabbit antibody (1 : 300, ab62215, Abcam, Cambridge, U.K.), anti-cav1 mouse antibody (1 : 500, 4H132, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), or anti-α-smooth muscle actin (SMA) mouse monoclonal antibody (1 : 1000, 1A4, Cell Signaling Technology, Danvers, MA, U.S.A.) at 4 °C for 12 h. After washing repeatedly, the cells were treated with Alexa 488-, Alexa 594- or Alexa 647-conjugated secondary antibodies (1 : 500, Thermo Fisher Scientific) at room temperature for 1 h. In some experiments, plasma membrane was stained with CF640-conjugated wheat germ agglutinin (WGA, 4 µg/mL in Hanks’ balanced salt solution, Biotium, Fremont, CA, U.S.A.) after fixation with 4% PFA for 10 min at 37 °C. Nuclei were stained with Hoechst 33342 (1 : 500, Thermo Fisher Scientific) at room temperature for 30 min.
Confocal ImagingConfocal images were obtained using a laser scanning confocal fluorescent microscope (A1R, Nikon, Tokyo, Japan). Fluorescence signals from GFP, secondary antibodies, and nuclear or plasma membrane indicators (Hoechst 33342 or CF640) were converted into binary images. Next, regions of interests (ROIs) corresponding to the fluorescence signals of (i) GFP within cytosol and nuclei (Figs. 1, 2), (ii) CaMK1α in plasma membrane, caveolae and nuclei (Fig. 3), and (iii) pCaMK1α within cytosol and nuclei (Fig. 4) were created by image arithmetic operations using the NIS Elements software (Nikon). The mean fluorescence intensity was calculated using these ROIs and ratios presented in the figures were obtained.
Ca2+ ImagingIntracellular Ca2+ imaging was performed using a confocal microscope (A1R). Cells were loaded with 10 µM Fluo4-AM (Thermo Fisher Scientific) for 15 min at room temperature. The excitation and emission wavelength were 488 nm and 500–530 nm, respectively. When the amplitude of [Ca2+]i elevation in primary ASMCs of cav1-KO and cav1-knockin cells were recorded, cells were loaded with 10 µM Fluo-4/AM and 20 µM Fura Red/AM for 30 min to perform ratiometric measurement.20) These indicators were excited at 488 nm and emission was collected at 500–530 nm (Fluo-4) and 662–737 nm (Fura-Red). FRatio was calculated by dividing fluorescence intensity of Fluo-4 by that of Fura-Red.
Real-Time PCRTotal RNA extraction, reverse transcription and real-time quantitative PCR were carried out as previously reported.20)
Data Notation and Statistical AnalysisPooled data sets are shown as the mean ± standard error of the mean (S.E.M.). The significance of differences between two groups was evaluated using the two-tailed t test after the application of the F test. Data from more than two groups were compared using one-way or two-way ANOVA followed by Tukey test. All statistical analysis was performed using BellCurve for Excel software (version 3.22; Social Survey Research Information, Tokyo, Japan). In all cases, p values <0.05 were considered to be significant. All data were obtained from at least three independent experiments.
DrugsDrugs were purchased from FUJIFILM Wako Pure Chemical Corporation except for nifedipine (MilliporeSigma), ethylene glycol-bis (2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA)-acetoxymethyl ester (AM) (Setareh Biotech, Eugene, OR, U.S.A.), 1,2-bis (2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA)-AM and HEPES (Dojindo, Kumamoto, Japan), and STO609 (Cayman Chemical, Ann Arbor, MI, U.S.A.). All hydrophobic compounds were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10–30 mM as a stock solution.
RESULTS
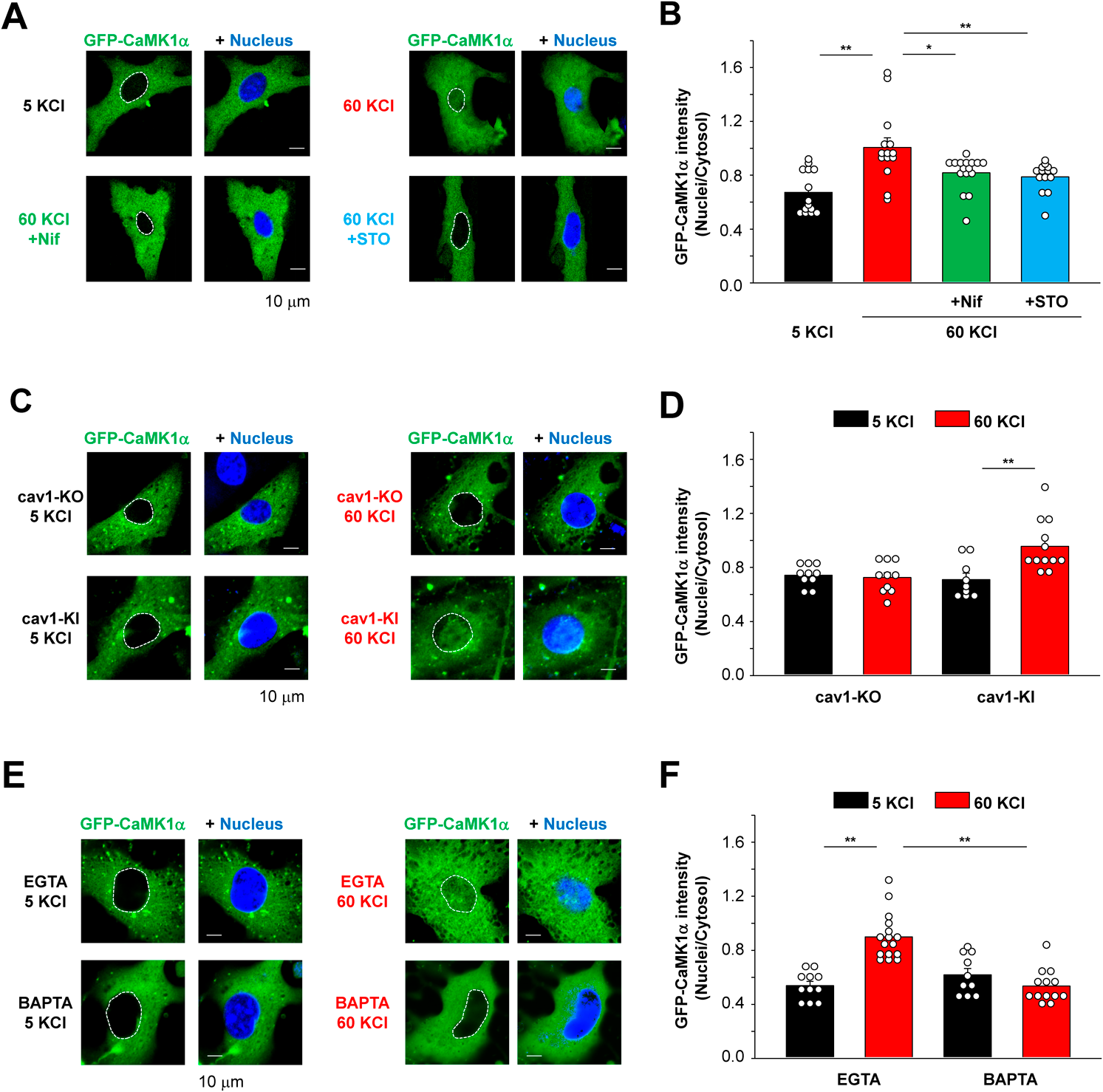
Live Cell Imaging Analysis Directly Demonstrates the Nuclear Translocation of CaMK1α in Primary ASMCsOur initial objective was to obtain direct evidence that depolarization-induced [Ca2+]i elevation can initiate CaMK1α translocation from cytosol to nucleus using a live cell imaging analysis. Since it is very difficult to express exogeneous proteins in freshly-isolated myocytes, we utilized unpassaged primary ASMCs.20) Immunostaining showed that these primary ASMCs were positive for a smooth muscle marker protein, SMA (Fig. 1A). Furthermore, these myocytes responded to a depolarizing stimulus (60 mM KCl solution) with a robust increase in [Ca2+]i (Fig. 1B). These findings strongly suggest that these primary ASMCs have Ca2+ signaling features that are similar to freshly-isolated vascular myocytes. These cells were induced to express GFP-tagged mouse CaMK1α using the adenovirus vector.20) As shown in Figs. 1C and D, consistent and abundant expression of GFP-CaMK1α was observed in primary ASMCs. In these experiments, the cells were depolarized using 60 mM KCl solution and the localization of GFP signals was continuously monitored using a confocal microscope. This sustained depolarizing stimulus elicited an increase in GFP-CaMK1α signals within the nucleus. A slight increase in GFP signals in the cytosol was also detected, probably because of myocyte contraction (Fig. 1C). On the other hand, the ratio of GFP-CaMK1α intensity within the nucleus to the cytosol continuously increased during depolarizing stimulus (Fig. 1C).
Next, we examined whether this nuclear translocation of GFP-CaMK1α can be reversed by switching the 60 mM KCl solution back to the 5 mM KCl solution. As shown in Fig. 1E, the depolarizing stimulus with 60 mM KCl for 20 min induced nuclear translocation of GFP-CaMK1α. However, membrane repolarization with 5 mM KCl solution did not relocate the intranuclear GFP-CaMK1α to the cytosol. These data convincingly suggest that CaMK1α can translocate to the nucleus upon membrane depolarization and subsequent [Ca2+]i elevation and that CaMK1α (probably phosphorylated CaMK1α) is stably localized in nucleus even though [Ca2+]i returns to basal levels (approximately 20 min after repolarization).
The Translocation of GFP-CaMK1α into the Nucleus Depends on Cav1.2, CaMKK2, and CaveolaeIn our previous study, we demonstrated that CaMK1α is activated by Ca2+ influx through Cav1.2 channels and that CaMK1α is phosphorylated by CaMKK2 within caveolae in primary ASMCs.20) In the present study, we further examined the contribution of Cav1.2, CaMKK2, and caveolae to the GFP-CaMK1α nuclear translocation. As shown in Figs. 2A and B, either the L-type Ca2+ channel blocker nifedipine (10 µM) or the CaMKK inhibitor STO609 (10 µM) inhibited the GFP-CaMK1α translocation to the nucleus. To evaluate the contribution of caveolae to the GFP-CaMK1α nuclear translocation, ASMCs isolated from cav1-KO mice were utilized. Previously, it has been demonstrated that the protein expression of cav1 and caveola structure were disrupted in smooth muscle tissues in cav1-KO mice,22) and that an increase in [Ca2+]i in cav1-KO myocytes due to membrane depolarization is equivalent to that in WT myocytes.20,23) Our results revealed that caveola disruption by cav1 gene deletion prevented depolarization-induced GFP-CaMK1α nuclear translocation, and that this was rescued by cav1-KI using the adenovirus vector20) (Figs. 2C, D). On the other hand, cav1-KI had no effects on 60 mM KCl-induced [Ca2+]i elevations and mRNA expression of CaMK1α and CaMKK2 in primary ASMCs isolated from cav1-KO mice (Supplementary Fig. S1).
It is well known that local Ca2+ signaling generated just beneath to the pores of neuronal Cav1 channels can cause intranuclear accumulation of phosphorylated CREB by Ca2+/CaM shuttling via CaMK.11,12) Our results also revealed that the local Ca2+ signaling within caveolae can induce CaMMK2 activation in primary ASMCs.20) This indicates that CaMK1α is phosphorylated by CaMKK2 at Ca2+ microdomains within caveolae and then translocates to the nucleus in vascular SMCs. To extend these findings, we utilized two types of Ca2+ chelators, EGTA-AM and BAPTA-AM.23,24) A fast chelator BAPTA can prevent local Ca2+ signaling but a slow chelator EGTA cannot. As shown in Fig. 2E, GFP-CaMK1α can translocate from the cytosol to the nucleus in myocytes treated with EGTA, but BAPTA inhibited this GFP-CaMK1α movement. Taken together, GFP-CaMK1α moves to the nucleus after Ca2+ influx through Cav1.2 and subsequent CaMKK2 activation within caveolae.
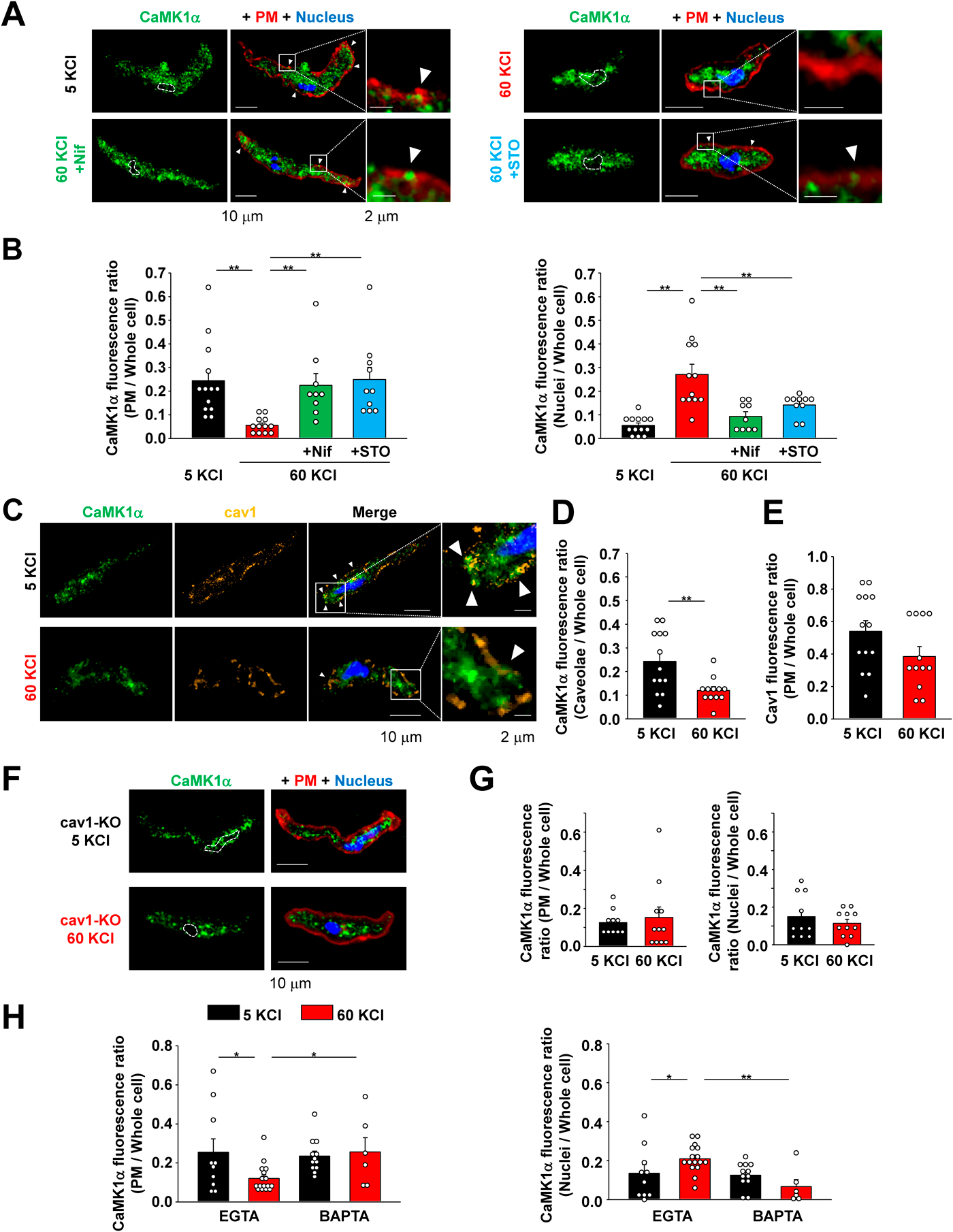
Depolarizing Stimulus Induces the Movement of CaMK1α from Caveolae to the Nucleus in Freshly-Isolated MASMCsAt present, it is unclear if endogenous CaMK1α can move from the cell surface to the nucleus in response to a depolarizing stimulus in freshly-isolated vascular SMCs. Therefore, we examined if CaMK1α is localized in the cell surface in resting state in freshly-isolated MASMCs, and also tested whether a depolarizing stimulus (using 60 mM KCl solutions) can produce translocation of CaMK1α to the nucleus. As shown in Fig. 3, at resting conditions, a part of CaMK1α signals were colocalized with the fluorescence signals from a plasma membrane marker, WGA. The depolarizing stimulus decreased the CaMK1α signals at the cell surface, while increasing the signals in the nucleus. This shift was inhibited by either nifedipine (10 µM) or STO609 (10 µM) (Figs. 3A, B).
This interesting pattern of results motivated us to study the spatial relationship between CaMK1α and caveolae. As shown in Fig. 3C, a portion of CaMK1α was localized in caveolae at resting conditions. This colocalization ratio was decreased upon membrane depolarization as shown in Figs. 3C and D. On the other hand, the same depolarizing stimulus did not modulate the cell surface cav1 level in freshly-isolated MASMCs (Fig. 3E). Intracellular localization of CaMK1α in freshly-isolated MASMCs from cav1-KO mice is illustrated in Figs. 3F and G. Note that the ratio of CaMK1α localized in the plasma membrane in cav1-KO myocytes was decreased compared to that in WT myocytes (Figs. 3B, G), and that no changes were detected after the depolarizing stimulus. It was also examined if a local Ca2+ elevation through Cav1.2 is sufficient to the CaMK1α nuclear translocation in freshly-isolated MASMCs using the two types of Ca2+ chelator (Fig. 3H). The fast Ca2+ chelator BAPTA inhibited the CaMK1α nuclear translocation, but the slow chelator EGTA showed no effects.
In combination, these data sets suggest that in freshly isolated myocytes a portion of CaMK1α is localized in caveolae via direct coupling with cav1 and that these CaMK1α molecules can move to the nucleus in response to Ca2+ influx and phosphorylation by CaMKK2 within caveolae.
Membrane Depolarization and Resulting Activation of Cav1.2 and CaMKK2 within Caveolae Causes Phosphorylation of CaMK1α in Freshly-Isolated MASMCsCaMK1α is known to be phosphorylated at Thr177 by its upstream kinase CaMKK and it then phosphorylates CREB at serine (Ser)133 to induce gene transcription.15) As mentioned above, it is unknown if the pCaMK1α translocates to the nucleus in response to membrane depolarization in freshly-isolated MASMCs. Accordingly, in our study, the phosphorylation of CaMK1α was visualized using a specific antibody to pCaMK1α. As shown in Fig. 4A, sustained membrane depolarizing stimulus increased pCaMK1α fluorescence intensity at both cytosol and nucleus. This phosphorylation was inhibited by the treatment with nifedipine or STO609 (Figs. 4A, B). In addition, CaMK1α phosphorylation could not be induced in the MASMCs isolated from cav1-KO mice (Figs. 4C, D). The phosphorylation of CaMK1α was also reduced by the fast chelator BAPTA, but not by the slow chelator EGTA (Fig. 4E).
These data provide evidence that Ca2+ influx through Cav1.2 and subsequent activation of CaMKK2 within caveolae leads to the phosphorylation of CaMK1α and pCaMK1α nuclear translocation in freshly-isolated MASMCs.
DISCUSSION
Relatively long-lasting membrane depolarization that produces [Ca2+]i elevation can induce E-T coupling in vascular myocytes.25,26) We have previously demonstrated that depolarization can induce transcription of genes related to chemotaxis, leukocyte adhesion and inflammation through the activation of a molecular complex consisting of Cav1.2/CaMKK2/CaMK1α that is located within caveolae of vascular myocytes.20) Results from this study implied that CaMK1α is fully activated at the cell surface and translocates to the nucleus, but direct evidence was not presented. In addition, it was unclear if CaMK1α actually translocates from the cell surface to the nucleus in freshly-isolated vascular myocytes. Based on the present study, the fluorescence imaging data and analysis provide the direct evidence that GFP-CaMK1α translocates from the cytosol to the nucleus in response to membrane depolarization, and subsequent [Ca2+]i elevation, as well as the activation of CaMKK2 within caveolae in primary ASMCs. Similar results were also obtained from freshly-isolated MASMCs (Figs. 3, 4). Taken together these results suggest that CaMK1α transport Ca2+ signals originated at the cell surface to the nucleus to induce E-T coupling.
CaMK1α Transduces Ca2+ Signals Generated at the Cell Surface to Nucleus in Vascular Smooth Muscle CellsPrevious studies have identified several different signal pathways that deliver Ca2+ signals from the cell surface to the nucleus. For example, in neurons, Ca2+ influx trough Ca2+ channels such as L-type Ca2+ channels and NMDA receptors in the plasma membrane activates specific downstream sensor molecules located close to the inner mouth of the channel pore.27) This then leads to the activation of transcription factors in the nucleus and gene induction for long-term synaptic plasticity.3) CaMK2γA transports Ca2+/CaM into the nucleus, resulting in CaMKK and CaMK4 activation.11) CaMK2γA contains classical nuclear localizing signal. It is inactive in the resting state, and dephosphorylation of Ser334 by calcineurin makes this signal effective in response to local Ca2+ elevation near L-type Ca2+ channel pores. In addition, CaMK1γ has been shown to play a similar role in parvalbumin-positive interneurons.12) We note, however, that mRNA of these CaMK molecules were not detectable in vascular smooth muscle.20) In contrast, in this study, we observed that CaMK1α is phosphorylated through the Cav1.2/CaMKK2 pathway, and that the phosphorylated CaMK1α can move to the nucleus, probably resulting in CREB phosphorylation.
It is known that CaMK1α shows cytosolic localization since CaMK1α has a classical NES at its C-terminus (312VVRHMRKLQL321).16) Chromosomal maintenance (CRM) 1 is responsible for CaMK1α nuclear export by binding this NES. On the other hand, translocation of CaMK1α into the nucleus depends on di-basic amino acid residues in the catalytic domain (263KR264).16) It has also been reported in murine lung epithelial cells that CaMK1α binds to NES in CTP:phosphocholine cytidylyltransferase (CCT) α and phosphorylates CCTα, which leads to the recruitment of 14-3-3ξ and subsequent nuclear entry.28) In this study, we observed that CaMK1α can translocate from the cell surface/caveolae to the nucleus after membrane depolarization. This translocation was dependent on Ca2+ influx through Cav1.2 channels and phosphorylation by CaMKK2 at caveolae (Fig. 2). This intranuclear localization of CaMK1α persisted even after [Ca2+]i returned to basal levels as has been reported in neurons.14) These results suggest that Ca2+/CaM binding, as well as phosphorylation by CaMKK2 at Thr177, produces conformational changes. These changes may promote nuclear translocation by (i) masking NES and dissociating CRM1 from CaMK1α, (ii) directly making the NLS effective, and (iii) recruiting binding partners such as 14-3-3ξ that trigger CaMK1α nuclear translocation. The sustained CaMK1α phosphorylation through these pathways may be sufficient to maintain intranuclear localization.
Interestingly, it has been reported that CaMK1γ can make homo-multimers in lipid rafts.10) Therefore, CaMK1α may form homo-multimers through its NES as in the case of CaMK1α-CCTα complex formation.28) This multimerization may mask NES and prevent the binding of CRM1.
CaMK1α Is Localized in Caveolae to Make a Molecular Complex with Cav1.2 and CaMKK2 in Vascular Smooth Muscle CellsCav1 is an essential component for caveola structure in smooth muscle tissues. It is also closely associated with various signaling pathway components, such as receptors, ion channels, transporters, and kinases, through caveolin-1 scaffolding domains.29,30) Our obtained data using myocytes from cav1-KO mice showed reduced CaMK1α localization in the plasma membrane. These results suggest that some portion of CaMK1α is tethered to the plasma membrane/caveolae through cav1. Notably, CaMK1α contains canonical cav1-binding motifs (235YEFPSPYW242). In addition, CaMK1γ is reported to be subject to prenylation followed by a kinase-activity-regulated palmitoylation.10) This type of modification is required for the targeting of CaMK1γ to lipid rafts in dendrites. Similar posttranscriptional modulation may be needed for CaMK1α to be localized in caveolae in vascular myocytes. The upstream molecules of CaMK1α, i.e., Cav1.2 and CaMKK2, also contain the canonical cav1-binding motif and are localized to caveolae.20) Our data demonstrate that CaMK1α, located within or very near caveolae, is fully activated in response to a prolonged depolarizing stimulus, and increase in [Ca2+]i; and that it then translocates into the nucleus where it phosphorylates CREB.
Our results raise a significant new question: Why can CaMK1α, localized only in caveolae, be phosphorylated and translocate to the nucleus after the activation of Cav1.2 and resulting global Ca2+ increases? Further studies are needed to resolve how CaMKK-mediated CaMK1α phosphorylation is controlled within or very near caveolae.
CONCLUSION
Our results strongly suggest that CaMK1α is phosphorylated at caveolae and then translocates to the nucleus in response to membrane depolarization in vascular smooth muscle. These new findings strengthen the working hypothesis that the activation of a molecular complex consisting of Cav1.2/CaMKK2/CaMK1α within caveolae can trigger the translocation of CaMK1α to the nucleus and cause CREB phosphorylation in vascular myocytes.
Acknowledgments
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 19H03381, 22H02773, and 21K19343 (to Y.S.), 17H05537, 19K07125 and 22H02787 (to H.Y.), and 18KK0218 (to Y.I.). We also acknowledge financial support through Grants-in-Aid for Research in Nagoya City University (1922007 to Y.S.) and Grant-in-Aid from Mitsui Sumitomo Insurance Welfare Foundation, Salt Science Research Foundation (Grant 1637), the Pharmacological Research Foundation, Tokyo, Suzuken Memorial Foundation, the Japan Foundation for Applied Enzymology, and the Uehara Memorial Foundation (to Y. S). We are grateful for the assistance the Research Equipment Sharing Center at Nagoya City University.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev., 96, 1261–1296 (2016).
- 2) Ferron L, Koshti S, Zamponi GW. The life cycle of voltage-gated Ca2+ channels in neurons: an update on the trafficking of neuronal calcium channels. Neurosignals, 5, NS20200095 (2021).
- 3) Woolfrey KM, Dell’Acqua ML. Coordination of protein phosphorylation and dephosphorylation in synaptic plasticity. J. Biol. Chem., 290, 28604–28612 (2015).
- 4) Ljubojevic-Holzer S, Herren AW, Djalinac N, et al. CaMKIIδC drives early adaptive Ca2+ change and late eccentric cardiac hypertrophy. Circ. Res., 127, 1159–1178 (2020).
- 5) Wamhoff BR, Bowles DK, McDonald OG, Sinha S, Somlyo AP, Somlyo AV, Owens GK. L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a Rho kinase/myocardin/SRF-dependent mechanism. Circ. Res., 95, 406–414 (2004).
- 6) Nystoriak MA, Nieves-Cintron M, Nygren PJ, Hinke SA, Nichols CB, Chen CY, Puglisi JL, Izu LT, Bers DM, Dell’acqua ML, Scott JD, Santana LF, Navedo MF. AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ. Res., 114, 607–615 (2014).
- 7) Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc. Natl. Acad. Sci. U.S.A., 105, 15623–15628 (2008).
- 8) Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. J. Biol. Chem., 279, 24064–24072 (2004).
- 9) Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron, 57, 94–107 (2008).
- 10) Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T, Hoshino M, Suzuki S, Kojima M, Mishina M, Okuno H, Bito H. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIγ. Neuron, 54, 755–770 (2007).
- 11) Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, Tsien RW. γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell, 159, 281–294 (2014).
- 12) Cohen SM, Ma H, Kuchibhotla KV, Watson BO, Buzsaki G, Froemke RC, Tsien RW. Excitation-transcription coupling in parvalbumin-positive interneurons employs a novel CaM kinase-dependent pathway distinct from excitatory neurons. Neuron, 90, 292–307 (2016).
- 13) Haribabu B, Hook SS, Selbert MA, Goldstein EG, Tomhave ED, Edelman AM, Snyderman R, Means AR. Human calcium-calmodulin dependent protein kinase I: cDNA cloning, domain structure and activation by phosphorylation at threonine-177 by calcium-calmodulin dependent protein kinase I kinase. EMBO J., 14, 3679–3686 (1995).
- 14) Schmitt JM, Guire ES, Saneyoshi T, Soderling TR. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J. Neurosci., 25, 1281–1290 (2005).
- 15) Wayman GA, Lee YS, Tokumitsu H, Silva AJ, Soderling TR. Calmodulin-kinases: modulators of neuronal development and plasticity. Neuron, 59, 914–931 (2008).
- 16) Stedman DR, Uboha NV, Stedman TT, Nairn AC, Picciotto MR. Cytoplasmic localization of calcium/calmodulin-dependent protein kinase I-alpha depends on a nuclear export signal in its regulatory domain. FEBS Lett., 566, 275–280 (2004).
- 17) Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron, 50, 897–909 (2006).
- 18) Uboha NV, Flajolet M, Nairn AC, Picciotto MR. A calcium- and calmodulin-dependent kinase Ialpha/microtubule affinity regulating kinase 2 signaling cascade mediates calcium-dependent neurite outgrowth. J. Neurosci., 27, 4413–4423 (2007).
- 19) Ageta-Ishihara N, Takemoto-Kimura S, Nonaka M, Adachi-Morishima A, Suzuki K, Kamijo S, Fujii H, Mano T, Blaeser F, Chatila TA, Mizuno H, Hirano T, Tagawa Y, Okuno H, Bito H. Control of cortical axon elongation by a GABA-driven Ca2+/calmodulin-dependent protein kinase cascade. J. Neurosci., 29, 13720–13729 (2009).
- 20) Suzuki Y, Ozawa T, Kurata T, Nakajima N, Zamponi GW, Giles WR, Imaizumi Y, Yamamura H. A molecular complex of Cav1.2/CaMKK2/CaMK1a in caveolae is responsible for vascular remodeling via excitation-transcription coupling. Proc. Natl. Acad. Sci. U.S.A., 119, e2117435119 (2022).
- 21) Kondo R, Kawata N, Suzuki Y, Yamamura H. Ca2+ signaling and proliferation via Ca2+-sensing receptors in human hepatic stellate LX-2 cells. Biol. Pharm. Bull., 45, 664–667 (2022).
- 22) Albinsson S, Shakirova Y, Rippe A, Baumgarten M, Rosengren BI, Rippe C, Hallmann R, Hellstrand P, Rippe B, Swärd K. Arterial remodeling and plasma volume expansion in caveolin-1-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol., 293, R1222–R1231 (2007).
- 23) Suzuki Y, Yamamura H, Ohya S, Imaizumi Y. Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+ (BKCa) and Cav1.2 Ca2+ channels and their clustering to regulate membrane excitability in vascular myocytes. J. Biol. Chem., 288, 36750–36761 (2013).
- 24) Fakler B, Adelman JP. Control of KCa channels by calcium nano/microdomains. Neuron, 59, 873–881 (2008).
- 25) Cartin L, Lounsbury KM, Nelson MT. Coupling of Ca2+ to CREB activation and gene expression in intact cerebral arteries from mouse : roles of ryanodine receptors and voltage-dependent Ca2+ channels. Circ. Res., 86, 760–767 (2000).
- 26) Pulver-Kaste RA, Barlow CA, Bond J, Watson A, Penar PL, Tranmer B, Lounsbury KM. Ca2+ source-dependent transcription of CRE-containing genes in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol., 291, H97–H105 (2006).
- 27) Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron, 16, 89–101 (1996).
- 28) Agassandian M, Chen BB, Pulijala R, Kaercher L, Glasser JR, Mallampalli RK. Calcium-calmodulin kinase I cooperatively regulates nucleocytoplasmic shuttling of CCTα by accessing a nuclear export signal. Mol. Biol. Cell, 23, 2755–2769 (2012).
- 29) Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol. Rev., 84, 1341–1379 (2004).
- 30) Dart C. Lipid microdomains and the regulation of ion channel function. J. Physiol., 588, 3169–3178 (2010).