Regular Articles
BET Inhibitors Induce p53-Independent Growth Arrest in HCT116 Cells via Epigenetic Control of the E2F1/c-MYC Axis
2023 年 46 巻 1 号 p. 12-18
詳細
2023 年 46 巻 1 号 p. 12-18
Although bromodomain and extraterminal (BET) inhibitors (BETis) have anti-tumor potential, the underlying molecular mechanism is poorly understood. We found that BETis effectively repressed cell growth via G1/S arrest and migration of HCT116 cells in a p53-independent manner. BETis increased the expression of p21WAF1 and repressed the expression of E2F target genes. Consistent with this, retinoblastoma protein (Rb) phosphorylation was downregulated by BETis, supporting E2F inactivation. To investigate the epigenetic mechanism, chromatin immunoprecipitation (ChIP) assays were employed using the E2F1 target gene c-MYC. Following BETi treatment, recruitment of phosphorylated Rb, BRD2, and MLL2 to the c-MYC promoter was reduced, whereas recruitment of unphosphorylated Rb and EZH2 was increased. Consequently, decreased H4K5/K12ac and H3K4me3 accumulation but increased H3K27me3 accumulation were observed. Overall, this study suggests that BETis may be useful for the treatment of colorectal cancer via epigenetic regulation of the E2F1/c-MYC axis, leading to growth arrest in a p53-independent manner.
Bromodomain and extraterminal (BET) proteins are transcriptional regulators that recognize acetylated lysine residues on histones via conserved tandem bromodomains 1 and 2.1,2) BET proteins comprise four subfamily proteins: BRD2, BRD3, BRD4, and BRDT.2) BET proteins modulate the transcription of genes involved in cell proliferation by recruiting transcriptional complexes.3) BET inhibitors (BETis), such as JQ-1 (pan-BRD inhibitor) and PFI-1 (BRD2- and BRD4-specific), have been designed to target the bromodomains of BET proteins.4,5) JQ-1 is the thienodiazepine class of BETis with similar binding affinities for all bromodomains of the BET family.6) In contrast, PFI-1 is a sulfonamide derivative which is highly selective for BRD2 and BRD4.7) Due to promising anti-cancer effects, diverse structural derivatives of BETi are under development.8,9) Recently, chiral analogues of PFI-1 at the sulfur core were synthesized for the treatment of myeloid leukemia.10) However, the molecular mechanism underlying BETi-mediated anti-cancer activity remains unclear.
An important tumor suppressor gene, TP53, is frequently mutated during tumor progression.11) As a transcriptional activator, p53 promotes the expression of p21WAF1, which is a cyclin-dependent kinase inhibitor, and induces cell cycle arrest.12) Another tumor suppressor, retinoblastoma protein (Rb), is dephosphorylated upon p21WAF1 activation. Rb hypo-phosphorylation leads to its association with E2F1, a transcription factor involved in cell cycle progression,13) and E2F1 inactivation via histone modifications.14–17) Although these tumor suppressors and BETis mediate anti-cancer functions, it is poorly explored whether p53 or Rb is required for the role of BETis in tumor suppression.
Using HCT116 cells, we evaluated the p53-independent activities of BETis, including growth arrest, p21WAF1 induction, and RB dephosphorylation. We also determined how BETis epigenetically modulate the expression of c-MYC by employing the transcription factor E2F1 and histone modifications.
The human colorectal cancer cell lines HCT116 wt (harboring wild-type (wt) p53) and p53-null HCT116 (HCT116 p53−/−), kindly provided by Professor Deug Y. Shin (Dankook University) with permission of Dr. Bert Vogelstein (Johns Hopkins University), were cultured in RPMI-1640 medium. Other cell lines used in this study were obtained from the American Type Culture Collection. HEK293T cells were cultured in Dulbecco’s modified Eagle medium. Polyethylenimine reagent (Polysciences Inc., Warrington, PA, U.S.A.) was used for transient transfection. In experiments using BETis, cells were exposed to 10 µM PFI-1 (Cayman Chemical, Ann Arbor, MI, U.S.A.) or 500 nM JQ1 (Merck Korea, Seoul, Korea). In the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, 1.5 × 103 cells per well were added to 96-well plates and treated with 50 µL MTT solution (2 mg/mL). In the clonogenic assay, 3 × 103 cells/well were added to 6-well plates and incubated for 7 d at 37 °C, after which the cells were stained with 0.05% crystal violet dye dissolved in distilled water. Cell migration assays were conducted at 80% confluence using a sterile yellow tip to induce a scratch, and the resulting images were analyzed using ImageJ software. For cell cycle analysis, cells were fixed with 80% (v/v) cold ethanol. Propidium iodide was added, and analysis was performed using the FACS Calibur flow cytometer and Cell Quest software (BD Biosciences, San Jose, CA, U.S.A.).
RNA Isolation and Real-Time Quantitative PCR (RT-qPCR)Total RNA was isolated from cells using TRIzol reagent (Thermo Fisher Scientific, Seoul, Korea) according to the manufacturer’s instructions, and 1 µg total RNA was reverse-transcribed using Moloney Murine Leukemia Virus Reverse Transcriptase (Thermo Fisher Scientific). Quantitative PCR was performed using the Icycler CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA, U.S.A.) and SYBR Green PCR mixture (Toyobo Co., Ltd., Osaka, Japan) as described previously.18) Primer sequences are provided in Supplementary Table 1. Expression levels were normalized to those glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an endogenous control.
Western BlottingFor Western blotting, cells were lysed in lysis buffer [50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, and 0.5% Nonidet P-40] supplemented with a protease inhibitor cocktail tablet (Roche, Mannheim, Germany). After sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), separated proteins were transferred to a polyvinylidene fluoride membrane, and the membranes were blocked in wash buffer containing 5% skim milk (BioBasic, San Diego, CA, U.S.A.). The membranes were then incubated with primary antibodies (Supplementary Table 2). After washing, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies. Protein bands were detected using the West-Zol substrate (iNtRON Biotechnology, Sungnam, Korea) and Vilber Fusion Solo 2 chemiluminescence system (Fisher Biotec, Wembley, Australia).
Immunoprecipitation (IP) and Chromatin Immunoprecipitation (ChIP)IP and ChIP were performed as described previously with some modifications.19) For IP, transfected HEK293T cells were lysed in radioimmunoprecipitation buffer. Cell lysates were incubated overnight at 4 °C with 1 µg antibody and protein A/G agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). After washing the beads with phosphate-buffered saline, the immune complexes were released from the beads by boiling and were analyzed by Western blotting. For ChIP, cells were fixed in 1% formaldehyde. Samples in SDS lysis buffer were sonicated using a Bioruptor (Tosho Denki, Yokohama, Japan). The supernatant was diluted five-fold in ChIP dilution buffer. Then, 1 µg antibody (Supplementary Table 3) and protein A/G agarose beads were used for immunoprecipitation. Immunoprecipitated chromatin complexes were uncrosslinked using a standard ChIP protocol. The DNA pellets were analyzed by qPCR using the following primer pair targeting the E2F1 binding site within the c-MYC promoter, producing a fragment of 130 bp20): AGGGCTTCTCAGAGGCTTG (forward) and TGCCTCTCGCTGGAATTACT (reverse).
Statistical AnalysisData are expressed as the mean ± standard deviation of at least three independent experiments. Comparisons between groups were performed using paired Student’s t-tests. p-Values < 0.05 (*), 0.01 (**), or 0.005 (***) were considered statistically significant.
To investigate the effects of BETis as anti-cancer agents and the correlation with p53 status, an MTT assay was performed using wt p53 and p53−/− HCT116 cells (Fig. 1A). Growth of these cells was significantly perturbed following treatment with PFI-1 (BRD2- and BRD4-specific) or JQ1 (pan-BRD-inhibitor) regardless of p53 status. Subsequent measurement of IC50 values supported p53-independent growth suppression by these BETis (Supplementary Fig. 1). Further clonogenic assays confirmed that the inhibitory effect of PFI-1 or JQ-1 on colony formation occurred regardless of p53 status (Fig. 1B). Overall, these data show that growth inhibition caused by PFI-1 or JQ-1 is p53 independent. Since the PFI-1 inhibitor is BRD2- and BRD4-specific, these may also play a role in HCT116 cells.
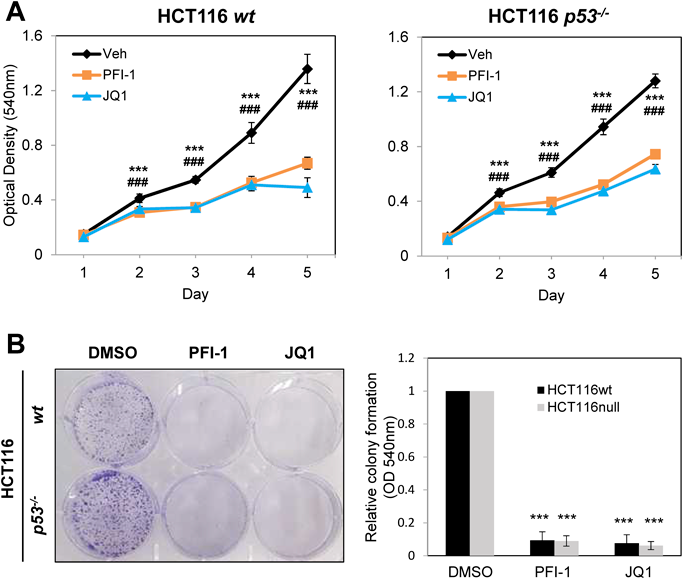
(A) Growth curves for HCT116 wt and p53−/− cells. Every 24 h, each well was treated with 50 µL MTT solution and incubated at 37 °C for 3 h. ***, p < 0.001 (Veh vs. PFI-1); ###, p < 0.001 (Veh vs. JQ1). (B) Clonogenic assay for HCT116 wt and p53−/− cells. On day 7, wells were stained with 0.05% crystal violet, and the absorbance of individual wells at 540 nm was measured (n = 3; ***, p < 0.001).
Since anti-cancer effects of BETis might also affect cell migration, a wound healing assay was performed using HCT116 wt and p53−/− cells. As shown in Fig. 2 (left), cell migration was significantly decreased following BETi treatment in both HCT116 wt and p53−/− cells. Further quantification of the covered area using ImageJ software demonstrated restricted cell migration by BETis and no apparent differences in the two cell lines (Fig. 2, right).

(A) Microscopy observations of wound healing assays in HCT116 wt and p53−/− cells. Cells were scratched prior to treatment with 10 µM PFI-1 or 500 nM JQ-1. 50× magnification. (B) Quantification of covered area, performed using ImageJ software (n = 3; ***, p < 0.001 compared with DMSO).
To investigate whether BETi-mediated suppression of cell proliferation and migration are associated with apoptosis, poly(ADP-ribose)polymerase (PARP) protein cleavage and nuclear fragmentation were monitored by Western blotting or Hoechst 33342 staining, respectively. As shown in Figs. 3A and B, neither PARP cleavage nor DNA fragmentation was detected. Next, we investigated the effect of BETis on cell cycle progression by propidium iodide staining (Supplementary Fig. 2). An increase in the G1 population was observed in BETi-treated cells regardless of p53 mutation; this was accompanied by a decrease in the S-phase cell population (Fig. 3C). The G2/M and sub-G1/G0 population were not affected by BETi treatment. Taken together, these data suggest that BETis suppress cancer properties, including cell proliferation and migration, but do not induce apoptosis, in a p53-independent manner.
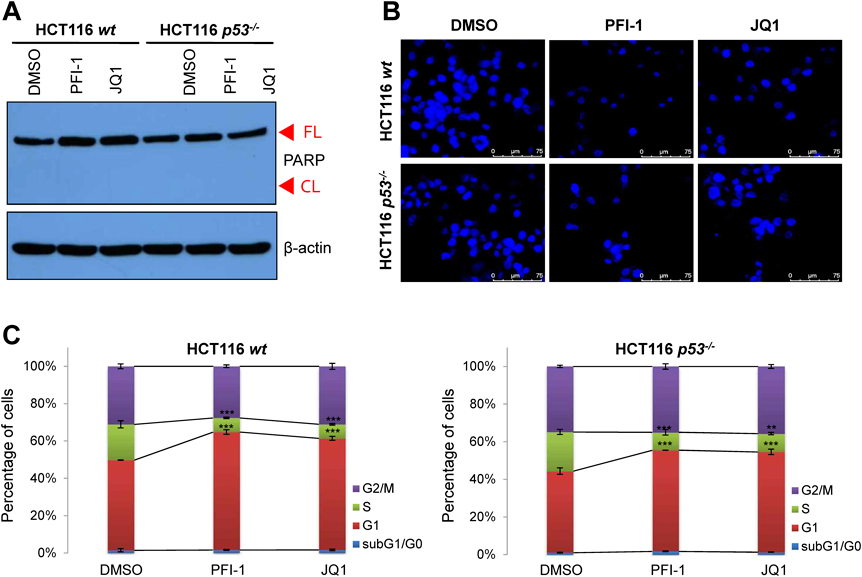
(A) Western blotting of cleaved PARP in HCT116 wt and p53−/− cells. FL, full length PARP protein; CL, cleaved PARP protein (cell death marker). (B) Nuclear staining using Hoechst 33342 to detect fragmented DNA. (C) Propidium iodide (PI) staining of HCT116 wt and p53−/− cells treated with PFI-1 (10 µM), JQ1 (500 nM), or vehicle (DMSO) for 24 h. After ethanol fixation, cells were stained with PI and analyzed using flow cytometry (n = 3; **, p < 0.01; ***, p < 0.001).
To analyze the molecular mechanism underlying BETi-induced G1 arrest, we first measured the mRNA expression of the p21WAF1 gene. RT-qPCR analysis indicated upregulation of p21WAF1 mRNA following treatment of both HCT116 cell lines with BETis (Fig. 4A). Consistent with this, elevated p21WAF1 protein was observed by Western blotting (Fig. 4B). As an inhibitor of cyclin-dependent kinases, p21WAF1 mediates dephosphorylation of the Rb protein.21) Therefore, we examined the level of Rb phosphorylation by Western blotting. As shown in Figs. 4C and D, BETis reduced Rb phosphorylation without affecting the overall level of Rb protein in both HCT116 cell lines; this may be a result of the BETi-dependent increase in p21WAF1. Overall, these data suggest that G1 arrest by BETis in HCT116 cells could be triggered by p21WAF1 induction followed by Rb dephosphorylation (or activation). The mechanism underlying p21WAF1 upregulation by BETi will require further investigation.

(A) RT-qPCR of p21 mRNA. Cells were treated with PFI-1 (10 µM), JQ-1 (500 nM), or DMSO for 6 h. RNA extraction and cDNA synthesis were performed according to standard protocols. (B) Western blotting to evaluate protein level of p21WAF1. (C) Cells were harvested 24 h after BETi treatment and subjected to Western blotting using antibodies against phospho-Rb (pRb), Rb, and β-actin (endogenous control). (D) Quantification of Rb phosphorylation. Phospho-Rb (pRb) intensity was measured by ImageJ software and normalized to that of pan-Rb bands (n = 3; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Next, we addressed how BETi-induced Rb dephosphorylation or activation regulates target gene expression via histone modification. As a transcription factor, E2F1, which is a well-known target of Rb activation, modulates the expression of genes associated with cell cycle progression and DNA replication.22) Dephosphorylated (active) Rb associates with E2F1 and represses its transcriptional activity.23) Interestingly, the BET member BRD2 interacts with E2F1 and stimulates expression of cyclin A, an E2F1-responsive gene.24) Our IP assay followed by Western blotting confirmed the interaction between E2F1 and BRD2 (Fig. 5A). BETi treatment induced downregulation of c-MYC, another target gene of E2F1, regardless of p53 status (Fig. 5B). We also examined downregulation of two other E2F1 target genes, E2F1 and ORC1L, in response to BETis (data not shown). For subsequent experiments, we used the c-MYC promoter, which harbors an E2F1-responsive element, and performed ChIP assays using antibodies against E2F1, BRD2, MLL2, H3K4me3, EZH2, and H3K27me3 following BETi treatment (Figs. 5C–H). The E2F1 binding specificity was determined using negative site (Supplementary Fig. 3). As expected, binding of E2F1 to the c-MYC promoter was not affected by BETi treatment (Fig. 5C). However, both PFI-1 and JQ-1 efficiently reduced BRD2 binding to the E2F1-responsive site in the c-MYC promoter (Fig. 5D). Accumulation of H4K5ac and H4K12ac, which are recognized by bromodomains of the BET family, was significantly reduced following BETi treatment (Supplementary Figs. 4A, B). We next investigated the effect of BETis on the chromatin deposition of histone H3 lysine 4 tri-methylation (H3K4me3), a histone modification catalyzed by the methyltransferase MLL family and required for transcriptional activation of E2F1.16) As shown in Fig. 5E, recruitment of MLL2 to the promoter was impaired by BETis in both types of HCT116 cells. Less H3K4me3 accumulation was observed in response to BETis (Fig. 5F). Conversely, we analyzed the effects of BETis on the accumulation of histone H3 lysine 27 tri-methylation (H3K27me3), a bivalent partner of H3K4me3. As a lysine-specific methyltransferase, EZH2 catalyzes H3K27 methylation, which represses PRC2 function.25) In contrast to MLL2, EZH2 binding to the promoter was increased in the presence of BETis (Fig. 5G), and thus chromatin occupancy of H3K27me3 was also enhanced (Fig. 5H). Overall, our ChIP data suggest that BETis reduce BRD2 binding to the c-MYC promoter without affecting E2F1 binding, leading to reduced chromatin occupancy of H4K5ac and H4K12ac. In addition, promoter deposition of bivalent H3K4me3 and H3K27me3 was inversely regulated in response to BETis. These differential chromatin occupancies following BETi treatment may result in transcriptional repression of the c-MYC gene.
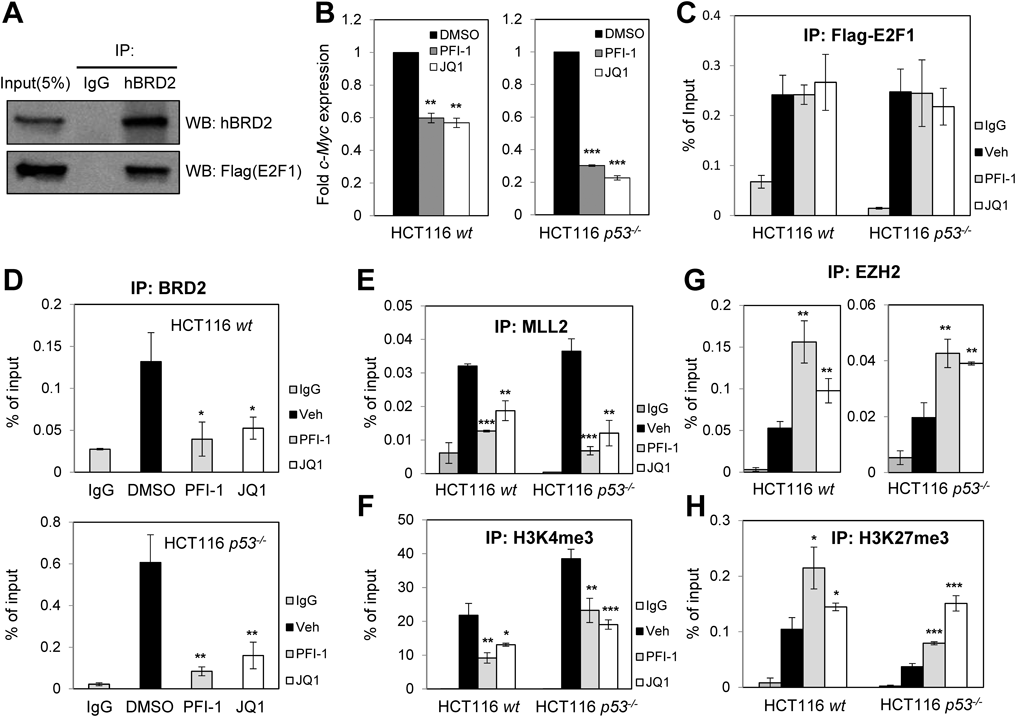
(A) Interaction between E2F1 and BRD2. HCT116 cells were transfected with Flag-E2F1 using PEI reagent. Cell lysates were subjected to immunoprecipitation using anti-BRD2 antibody (use IgG as a control antibody), and precipitated proteins were visualized by Western blotting using anti-BRD2 and anti-Flag antibodies. (B) Downregulation of c-MYC mRNA by BETis. Cells were treated with PFI-1 (10 µM), JQ-1 (500 nM), or DMSO for 6 h. (C, D) Recruitment of E2F1 and BRD2 to the c-MYC promoter. ChIP assays using anti-Flag (E2F1) or anti-BRD2 were performed 24 h after BETi treatment (10 µM PFI-1 or 500 nM JQ-1). (E–H) Epigenetic regulation of the c-MYC promoter by BETi. ChIP assays were performed using antibodies against MLL2 (E), H3K4me3 (F), EZH2 (G), or H3K27me3 (H) following BETi treatment. The c-MYC promotor region including the E2F1-responsive site was amplified and analyzed by qPCR. (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
In this study, we investigated the molecular and epigenetic mechanisms underlying the effects of the BETis PFI-1 and JQ1 as potential anti-cancer agents. BETi treatment attenuated proliferation and migration of HCT116 colon cancer cells in a p53-independent manner and induced G1 arrest, but with no effect on apoptosis. Upregulation of p21WAF1 and thereby Rb dephosphorylation may account for the BETi-induced G1 arrest. Rb dephosphorylation leads to tight Rb binding to the transcription factor E2F1, which recruits histone-modifying enzymes for transcriptional repression.13–17) Moreover, ChIP assays revealed that the epigenetic mechanism underlying BETi-mediated E2F1 inactivation involved the E2F1 binding site of the c-MYC promoter. Most importantly, BETis induced inverse effects on deposition of the bivalent histones H3K4me3 and H3K27me3 at the c-MYC promoter. Collectively, our data suggest that BETis act as anti-cancer agents by indirectly inducing p21WAF1 and directly regulating the Rb/E2F1 axis associated with diverse histone modifications.
However, some questions remain. How do BETis upregulate p21WAF1 in a p53-independent manner? mRNA expression of p21WAF1 could be regulated independently of p53.26) Previous studies have provided molecular insight into p53-independent regulation of p21WAF1 in the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway or at DNA damage checkpoints.27,28) At the transcriptional level, transcription factors such as FOXO1/3 and ZNF84 have been reported to be p53-independent activators of p21WAF1.29,30) Since BET proteins are transcriptional co-regulators that may interact with transcription factors to modulate gene activity, identification of such transcription factors will be important to understand p53-independent transcriptional activation of p21WAF1 by BETis. Interestingly, it has been reported that YAP, a transcriptional repressor of p21WAF1, is downregulated upon JQ1 treatment, leading to p53-independent p21WAF1 upregulation and subsequent c-MYC downregulation for growth repression in chondrosarcoma cells.31) In terms of epigenetics, reduced activity of histone deacetylases by Trichostatin A (TSA) resulted in decreased p53-independent p21WAF1 expression.32) As BET proteins recognize acetylated histone lysine residues, the role of these residues in histone tails needs further investigation to improve our understanding of the epigenetic regulation of p21WAF1. To investigate the effects of BETis on p21WAF1, we are currently mapping the responsible p21WAF1 promotor sites (data not shown). It will also be important to determine which histone deacetylases deacetylate H4K5ac or H4K12ac following BETi treatment. Future studies will aim to improve our knowledge of the epigenetic role of BET proteins and BETis.
This study was supported in part by a Grant of the Basic Science Research Program (NRF-2020R1A2C1007445), and a Grant of the Bio & Medical Technology Development Program (NRF-2018M3A9H1023139) funded by the Korean government, MSIT.
The authors declare no conflict of interest.
This article contains supplementary materials.