Abstract
The negative inotropic effects of nine Vaughan Williams class I antiarrhythmic drugs were examined in guinea pig ventricular tissue preparations. The drugs decreased the contractile force of papillary muscles with different potencies: the potency order was propafenone > aprindine > cibenzoline > flecainide > ranolazine > disopyramide > pilsicainide > mexiletine > GS-458967. The potency of drugs correlated with the reported IC50 values to block the L-type Ca2+ channel rather than the Na+ channel. The effects of drugs were roughly the same when examined under a high extracellular K+ solution, which inactivates the Na+ channel. Furthermore, the attenuation of the extracellular Ca2+-induced positive inotropy was strong with propafenone, moderate with cibenzoline, and weak with pilsicainide. These results indicate that the negative inotropic effects of class I antiarrhythmic drugs can be largely explained by their blockade of the L-type Ca2+ channel.
INTRODUCTION
Vaughan Williams Class I antiarrhythmic drugs have been used in the pharmacological treatment of arrhythmia; they inhibit the propagation of ectopic excitation through the blockade of Na+ channels.1) Class I antiarrhythmic drugs are used differently depending on their mode of action on the Na+ channel and other pharmacological effects via receptors, channels, and transporters.1) Although class I antiarrhythmic agents are moderately effective against various types of arrhythmia, they often show unwanted cardiosuppressive effects that limit their use in patients with reduced cardiac function.2,3) On the other hand, the development and clinical use of class I antiarrhythmic drugs with a novel mode of action on the Na+ channel are now in progress.1,4) For the optimization of the clinical usage of class I antiarrhythmic drugs and the development of new antiarrhythmic drugs with higher efficacy and safety, understanding the mechanism of their cardiosuppressive effects is essential. In this study, we compared the negative inotropic effects of nine class I antiarrhythmic drugs using isolated guinea pig ventricular myocardium to obtain a birds-eye overview concerning the factors underlying their negative inotropic effects.
MATERIALS AND METHODS
All experiments were performed in accordance with the Guiding Principles for the Care and Use of Laboratory Animals approved by The Japanese Pharmacological Society and the Guide for the Care and Use of Laboratory Animals at the Faculty of Pharmaceutical Sciences, Toho University (22-41-507).
The negative inotropic effects of nine class I antiarrhythmic drugs and related agents were examined in isolated right ventricular papillary muscles from Hartley strain male guinea pigs. The procedures were the same as those in our previous study.5,6) The papillary muscles were mounted in an organ bath filled with a physiological salt solution of the following composition: 118.4 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 24.9 mM NaHCO3, and 11 mM glucose (pH = 7.4, 37 °C), gassed with 95% O2−5% CO2 and maintained at 36 ± 0.5 °C. The preparations were electrically driven at 0.5 Hz, and the contractile force was recorded isometrically. High extracellular K+ solution contained 118.4 mM NaCl, 30 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 24.9 mM NaHCO3, 11 mM glucose, and 1 µM (S)-(−)-Bay K8644 (pH = 7.4, 37 °C). Small aliquots of drug solutions were added to the solution in the organ bath to obtain the desired final concentrations.
Tetrodotoxin citrate and pilsicainide were purchased from Alomone Labs (Jerusalem, Israel), GS-458967 from MedKoo Biosciences (Morrisville, NC, U.S.A.), disopyramide from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), cibenzoline and aprindine from Cosmo Bio Co., Ltd. (Tokyo Japan), nifedipine, flecainide, mexiletine, and (S)-(−)-Bay K8644 from Sigma-Aldrich (St. Louis, MO, U.S.A.), propafenone from LKT Laboratories, Inc. (St. Paul, MN, U.S.A.), and ranolazine from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). GS-458967, nifedipine, disopyramide, flecainide, propafenone, ranolazine, and (S)-(−)-Bay K8644 were dissolved in dimethyl sulfoxide and other chemicals in distilled water.
All data were expressed as mean ± standard error of the mean (S.E.M). IC50 values were calculated by non-linear regression analysis using GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA, U.S.A.). Pearson’s coefficients were used for evaluating correlations between the IC50 for the negative inotropy and the IC50 for Na+ or Ca2+ channel blockade.
RESULTS
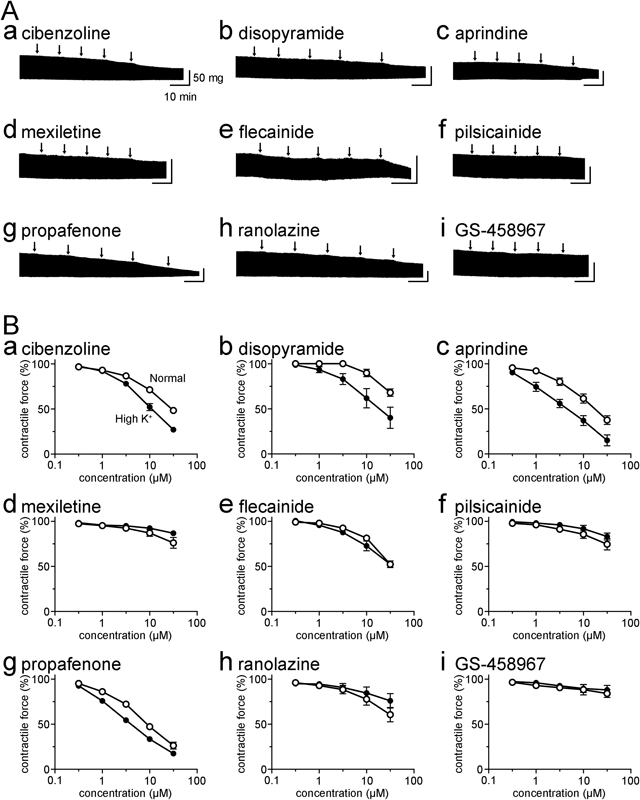
The class I antiarrhythmic drugs decreased the contractile force of the right ventricular papillary muscles (Fig. 1). The effects were dependent on the concentration of the drugs ranging from 0.3 to 30 µM; the concentration range includes or largely overlaps with the therapeutic plasma concentration of the drugs.7,8) There was a large difference in the potency of drugs ranging from propafenone, which showed a marked reduction of contractile force, to GS-458967, which showed virtually no effect. The potency order was propafenone > aprindine > cibenzoline > flecainide > ranolazine > disopyramide > pilsicainide > mexiletine > GS-458967. The contractile force in the presence of 30 µM of each drug expressed as a percentage of that in their absence was 26.3 ± 3.8% (n = 5) for propafenone, 37.5 ± 4.7% (n = 5) for aprindine, 48.4 ± 3.1% (n = 5) for cibenzoline, 52.3 ± 2.4% (n = 5) for flecainide, 60.6 ± 8.0% (n = 5) for ranolazine, 68.1 ± 4.1% (n = 5) for disopyramide, 74.7 ± 6.4% (n = 5) for pilsicainide, 76.2 ± 6.0% (n = 5) for mexiletine, and 84.1 ± 4.4% (n = 5) for GS-458967.
The effects of class I antiarrhythmic drugs were also examined under elevated extracellular K+ concentration, under which the function of Na+ channels is markedly reduced while the dependence of myocardial contraction on the L-type Ca2+ channel is maintained9,10); this was confirmed by the observation that the contractile force was highly sensitive to nifedipine but not to tetrodotoxin. The contractile force under elevated extracellular K+ concentration was decreased to 10.7 ± 3.9% (n = 5) with 1 µM nifedipine, but to 89.1 ± 3.0% (n = 5) with 10 µM tetrodotoxin. The effect of class I antiarrhythmic drugs was not strongly affected by elevated extracellular K+ concentration, and the overall trend was the same as that under normal extracellular solution (Fig. 1).
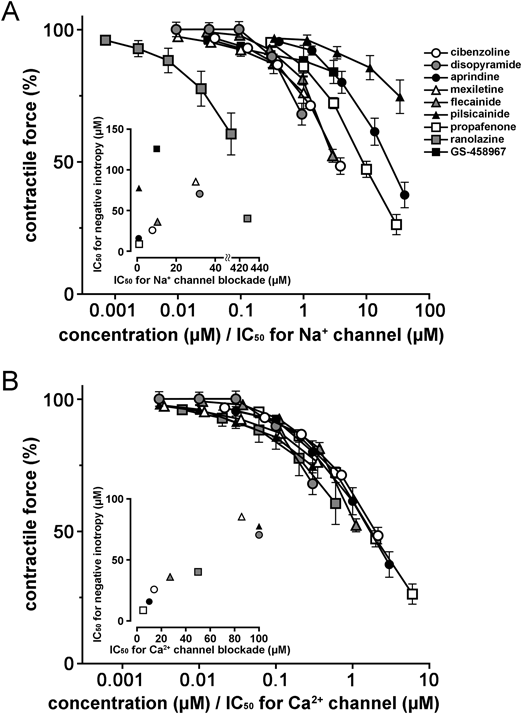
To clarify the ionic mechanisms for the negative inotropic effects of class I antiarrhythmic drugs, we analyzed our data using the reported potency of drugs against the Na+ and Ca2+ channels (Supplementary Table S1). When the concentration of each drug was expressed as a ratio against its IC50 value for Na+ channel, there was about a 1000-fold difference in the potency (Fig. 2A), and the IC50 for negative inotropy of drugs did not correlate with the IC50 for Na+ channel (r=−0.10, p = 0.80). In contrast, when the concentration of each drug was expressed as a ratio against its IC50 value for Ca2+ channel, the negative inotropic effects of all drugs tended to converge on a single concentration–response curve; the difference in potency was decreased to 10-fold (Fig. 2B). There was a correlation between the IC50 for negative inotropy in the normal solution and the IC50 for Ca2+ channel (r = 0.96, p = 0.0001).
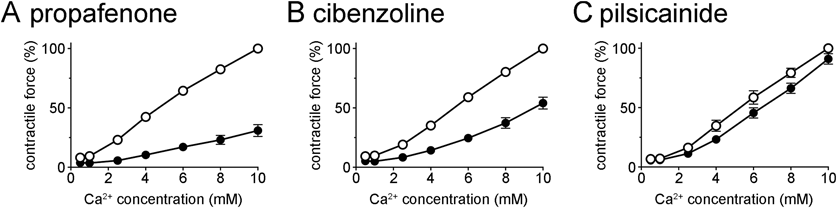
To further confirm the involvement of Ca2+ channel blockade, the effects of drugs on the inotropic response to extracellular Ca2+ were examined. The extracellular Ca2+ concentration was increased up to 10 mM in the absence and presence of drugs. The inotropic response was markedly decreased by nifedipine, which confirms the Ca2+ channel-dependent nature of this response; the inotropic response to 10 mM Ca2+ in the presence of 0.3 µM nifedipine was 33.1 ± 3.6% (n = 5) of that in its absence. The class I antiarrhythmic drugs showed inhibitory effects on this response. The potency order of inhibition was propafenone > cibenzoline > pilsicainide. The response to 10 mM Ca2+ in the presence of 30 µM propafenone, cibenzoline, and pilsicainide was 30.9 ± 5.0% (n = 5), 54.0 ± 5.0% (n = 5), and 91.1 ± 4.4% (n = 5) of that in its absence, respectively.
DISCUSSION
In the present study, we systematically examined the negative inotropic effects of nine class I antiarrhythmic drugs in isolated myocardial preparations to obtain a birds-eye overview concerning the factor(s) underlying their cardio-suppressive effects (Fig. 1). The results revealed a large difference in the negative inotropic effect of drugs. The potency order was propafenone > aprindine > cibenzoline > flecainide > ranolazine > disopyramide > pilsicainide > mexiletine > GS-458967. The present results with the class Ia and Ib antiarrhythmic drugs, cibenzoline, disopyramide, aprindine, and mexiletine, were consistent with earlier reports.2,11)
The negative inotropic effects of class I antiarrhythmic drugs have been attributed to the Na+ blocking effects themselves. A plausible explanation was the involvement of the Na+-Ca2+ exchanger; Na+ channel blockade decreases intracellular Na+ concentration, causing a compensatory intracellular uptake of Na+ via Na+-Ca2+ exchanger and an extracellular efflux of Ca2+, which results in a reduction of contractile force.12) However, the observed large difference in the negative inotropic potency among drugs raises the possibility that Na+ channel blockade itself is not the major mechanism for the negative inotropy. In fact, the large variation among drugs was not resolved even when the concentration of drugs was expressed as a ratio against their IC50 value for Na+ channel blockade (Fig. 2A). Experimentally, the negative inotropic effects of drugs were not much affected by the elimination of Na+ channel function by elevated extracellular K+ (Fig. 1). These results clearly indicated that factors other than Na+ channel blockade are largely involved in the negative inotropic action of class I antiarrhythmic drugs.
Trans-sarcolemmal Ca2+ influx through Ca2+ channels is known to be the main trigger of myocardial contraction; it triggers Ca2+ release from the sarcoplasmic reticulum to form the intracellular Ca2+ transient. The myocardial contractile force under elevated extracellular K+ concentration, used in the present study, was shown to be highly dependent on Ca2+ channel function.9,10) The present observation that the negative inotropic effect of class I antiarrhythmic drugs observed under normal conditions was mostly preserved under elevated extracellular K+ conditions implies the involvement of Ca2+ channel blockade. The negative effects of all drugs tended to converge on a single concentration–response curve when the concentration of each drug was expressed as a ratio against its IC50 value for Ca2+ channel blockade (Fig. 2B). Furthermore, there was a strong correlation between the negative inotropic effects of class I antiarrhythmic drugs and Ca2+ channel blockade (r = 0.96, p = 0.0001). These results indicated that the negative inotropic action of class I antiarrhythmic drugs is mostly due to their Ca2+ channel blocking activity. This was further confirmed by the observation that the inhibitory effects of propafenone, cibenzoline, and pilsicainide on the extracellular Ca2+-induced inotropy correlated with their potency of negative inotropic effects (Fig. 3).
The negative inotropic effects of class I antiarrhythmic drugs do not appear to correlate with the Vaughan–Williams classification based on their effects on action potential duration (APD).1) Class Ia antiarrhythmic drugs, cibenzoline and disopyramide, prolong APD through K+ channel blockade; this might cancel their negative inotropic effects through extension of the time for trans-sarcolemmal Ca2+ influx through Ca2+ channels. This was not the case in the present results, which implies that the extension of channel opening could not overcome the channel blockade. Class Ib antiarrhythmic drugs, aprindine and mexiletine, are known to shorten ventricular APD, which may result in negative inotropy through reduction of Ca2+ influx. As the effects of these drugs are frequency dependent, the lack of negative inotropy under the present experimental condition may be due to the low stimulation frequency (0.5 Hz). Class Ic antiarrhythmic drugs, flecainide, pilsicainide, and propafenone, dissociate from the Na+ channel slowly and results in a potent blockade of the channel. The present results revealed a large variation in the negative inotropic potency, suggesting that factors other than Na+ channel blockade are the determinants of negative inotropy. Class Id is a newly adopted subclassification defined as blockers of the persistent component of the Na+ channel current referred to as late INa.1) Ranolazine and GS-458967, which belong to class Id, showed moderate and weak negative inotropy, respectively. Ranolazine was reported to have additional sites of action including the sarcoplasmic reticulum Ca2+ release channel.13) On the other hand, GS-458967 and NCC-3902, highly selective blockers of late INa, were reported to decrease the contractile force of isolated myocardium only slightly.4,14) It appears that selective blockade of late INa causes only a weak negative inotropy.
Some limitations of the present study, both in the experimental design and extrapolation of the results to cardiac function in vivo, should be mentioned. Firstly, we used a single experimental protocol for all class I antiarrhythmic drugs to make a fair comparison of their negative inotropic effects, but considering that these drugs have different modes of action such as voltage and/or frequency dependence, the obtained potency order may not be absolute. In this sense, we are caught in a dilemma. Secondly, we used a high K+ extracellular solution to eliminate the contribution of the Na+ channel, but the possibility that the remaining contractile mechanisms were affected cannot be excluded. For example, the negative inotropic effects of aprindine, cibenzoline, and disopyramide were enhanced under high K+ condition; the mechanisms underlying this phenomenon are unclear at present. Finally, we used field-stimulated papillary muscles to measure the negative inotropic effects of class I antiarrhythmics free from their effects on conduction. In the whole heart, however, a decrease in conduction velocity itself may cause a decrease in cardiac output through desynchronization of contraction among various regions of the ventricular wall.15) Thus, to evaluate the cardiosuppressive effects of class I antiarrhythmic drugs comprehensively, their effects on conduction must also be incorporated.
In conclusion, the present study showed that the negative inotropic effects of class I antiarrhythmic drugs correlate not with the blockade of the Na+ channel but with the blockade of the Ca2+ channel. Thus, novel blockers of the Na+ channel with minimum cardiosuppression can probably be developed, provided that they have no Ca2+ channel blocking activity.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Numbers JP20K16013, JP20K07299, and JP20K07091. H.H. received Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (N-201503).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Lei M, Wu L, Terrar DA, Huang CL. Modernized classification of cardiac antiarrhythmic drugs. Circulation, 138, 1879–1896 (2018).
- 2) Nawada T, Tanaka Y, Hirai S, Hisatome I, Hasegawa J, Kotake H, Mashiba H. Evaluation of negative inotropic and antiarrhythmic effects of class 1 antiarrhythmic drugs. Int. J. Clin. Pharmacol. Ther., 32, 347–355 (1994).
- 3) JCS Joint Working Group. Guidelines for Pharmacotherapy of Atrial Fibrillation (JCS 2013). Circ. J., 78, 1997–2021 (2014).
- 4) Irie M, Hiiro H, Hamaguchi S, Namekata I, Tanaka H. Involvement of the persistent Na+ current in the diastolic depolarization and automaticity of the guinea pig pulmonary vein myocardium. J. Pharmacol. Sci., 141, 9–16 (2019).
- 5) Hamaguchi S, Kariya M, Ozaki AF, Namekata I, Tanaka H. Contribution of ATP-mediated positive feedback to sympathetic nerve-induced positive inotropy in Guinea pig ventricular myocardium. Biol. Pharm. Bull., 44, 458–460 (2021).
- 6) Hamaguchi S, Abe K, Komatsu M, Kainuma J, Namekata I, Tanaka H. Positive lusitropic effect of quercetin on isolated ventricular myocardia from normal and streptozotocin-induced diabetic mice. Biol. Pharm. Bull., 44, 1894–1897 (2021).
- 7) Tamura A, Ogura T, Uemura H, Reien Y, Kishimoto T, Nagai T, Komuro I, Miyazaki M, Nakaya H. Effects of antiarrhythmic drugs on the hyperpolarization-activated cyclic nucleotide-gated channel current. J. Pharmacol. Sci., 110, 150–159 (2009).
- 8) Yonemizu S, Masuda K, Kurata Y, Notsu T, Higashi Y, Fukumura K, Li P, Ninomiya H, Miake J, Tsuneto M, Shirayoshi Y, Hisatome I. Inhibitory effects of class I antiarrhythmic agents on Na+ and Ca2+ currents of human iPS cell-derived cardiomyocytes. Regen. Ther., 10, 104–111 (2019).
- 9) Hirth C, Borchard U, Hafner D. Effects of the calcium antagonist diltiazem on action potentials, slow response and force of contraction in different cardiac tissues. J. Mol. Cell. Cardiol., 15, 799–809 (1983).
- 10) Kondo N, Mizukami M, Shibata S. Negative inotropic effects of disopyramide on guinea-pig papillary muscles. Br. J. Pharmacol., 101, 789–792 (1990).
- 11) Honerjäger P, Loibl E, Steidl I, Schönsteiner G, Ulm K. Negative inotropic effects of tetrodotoxin and seven class 1 antiarrhythmic drugs in relation to sodium channel blockade. Naunyn Schmiedebergs Arch. Pharmacol., 332, 184–195 (1986).
- 12) Ito K, Nagafuchi K, Taga A, Yorikane R, Koike H. Possible involvement of altered Na+-Ca2+ exchange in negative inotropic effects of class I antiarrhythmic drugs on rabbit and rat ventricles. J. Cardiovasc. Pharmacol., 27, 355–361 (1996).
- 13) Parikh A, Mantravadi R, Kozhevnikov D, Roche MA, Ye Y, Owen LJ, Puglisi JL, Abramson JJ, Salama G. Ranolazine stabilizes cardiac ryanodine receptors: a novel mechanism for the suppression of early afterdepolarization and torsades de pointes in long QT type 2. Heart Rhythm, 9, 953–960 (2012).
- 14) Namekata I, Hiiro H, Odaka R, Saito T, Hamaguchi S, Tsukamoto T, Ishikawa R, Katayama Y, Kondo Y, Tanaka H. Inhibitory effect of a late sodium current blocker, NCC-3902, on the automaticity of the guinea pig pulmonary vein myocardium. Biol. Pharm. Bull., 45, 1–10 (2022).
- 15) Rabêlo Evangelista AB, Monteiro FR, Nearing BD, Belardinelli L, Verrier RL. Flecainide-induced QRS complex widening correlates with negative inotropy. Heart Rhythm, 18, 1416–1422 (2021).