Abstract
Peripheral neuropathy is one of the major adverse effects that limit the clinical application of bortezomib (BTZ). However, the underlying mechanisms of BTZ-induced peripheral neuropathy (BIPN) remain elusive. To examine cell types potentially involved in the development of BIPN, we used four purified cultures of cells of the peripheral nervous system: Schwann cells (SCs), satellite glial cells (SGCs), macrophages, and dorsal root ganglion (DRG) neurons. Administration of a low BTZ concentration (5 nM; similar to concentrations in clinical use) caused dedifferentiation of cultured SCs, returning mature SCs to an immature state. In cultured SGCs, BTZ increased glial fibrillary acidic protein (GFAP) levels without inducing the release of inflammatory cytokines or chemokines. In macrophages, BTZ caused little inflammatory response. Finally, in DRG neurons, BTZ strongly suppressed the expression levels of sensor and transducer ion channels without affecting cell morphology. Taken together, low concentrations of BTZ can cause SC dedifferentiation (i.e., demyelination), increased GFAP level in SGC, and decreased expression levels of sensor and transducer ion channels in DRG neurons (i.e., numbness feeling). Thus, we have reported, for the first time, specific effects of BTZ on peripheral nervous system cells, thereby contributing to a better understanding of the initiating mechanism of BIPN.
INTRODUCTION
Bortezomib (BTZ) is a small-molecule anti-cancer agent and reversible proteasome inhibitor that targets the β5 subunit of the 20S proteasome.1) BTZ can terminate ubiquitination-mediated protein degradation and cause severe protein overload in metabolically active cells.2) With this, BTZ can suppress the proliferation of cancer cells and has thus been widely utilized in the clinical treatment of multiple myeloma.3) However, approximately 30% of patients exhibit peripheral neuropathy, including both positive (e.g., pain, burning, and tingling) and negative (e.g., numbness, sensory loss, and glove anesthesia) symptoms.4) These adverse effects are often unendurable and usually lead to dose limitation or even treatment termination. In some extreme cases, the QOL of patients is even permanently damaged.5) Therefore, the pathogenesis of BTZ-induced peripheral neuropathy (BIPN) must be elucidated; however, little is known.
The peripheral nervous system consists of two major parts: the somatic and autonomic nervous systems. The sensory system is included in the former. Generally, sensory disorders are the most widely researched peripheral neuropathies because they are easy to validate by behavioral tests on rodent models. The sensory system consists of afferent nerves and dorsal root ganglions. Afferent nerves include myelinated axons with middle- or large-diameter neuronal somas (myelinated by Schwann cells (SCs)) and unmyelinated axons with small-diameter neuronal somas. Dorsal root ganglions (DRGs) consist of the three above-mentioned types of neurons, which are further enveloped and supported by satellite glial cells (SGCs) and resident macrophages. The central terminal of DRG neurons projects to the dorsal horns of the spinal cord, and its signals are modified and transmitted by microglia and astrocytes. The working of the sensory system requires all these components to function appropriately. Because BTZ induces peripheral neuropathy, it is assumed to have an adverse effect on the sensory system4); however, few studies have investigated possible causes at the cellular level.
Neuronal inflammation and degeneration are common features in BIPN models, characterized by macrophage infiltration at both sciatic nerves and DRGs and immune activation of SGCs at DRG sites.6) The capability of BTZ to cross healthy blood-brain barriers is limited, and thus the activation of glial cells in the central nervous system is recognized as a subsequent outcome of peripheral nerve damage.6–9) Furthermore, pathological changes in peripheral nervous systems have also been reported in BIPN models and are characterized by damaged myelin sheaths and degenerated nerve endings.10–12) Furthermore, BTZ treatment also causes vacuolation in SGCs.13) Published research cannot provide clear interpretations on the pathogenesis of BIPN because in vivo models are mostly validated in chronic behavior models, which cannot illustrate the initial phase of neuropathy development. The convergence of downstream phenotypes as neuronal inflammation may obscure the original cytotoxic features of BTZ. Thus, our current study used purified primary cell cultures (of peripheral glial cells (SCs and SGCs), macrophages, and DRG neurons) to eliminate inter-cellular signal crosstalk and reveal the direct impact of BTZ on each element of the peripheral sensory system at an acute stage.
MATERIALS AND METHODS
AnimalsAll animal care and experimental procedures were in accordance with the ethical guidelines of the Kyoto University Animal Research Committee. C57BL/6J mice and Wistar/ST rats were purchased from Japan SLC (Shizuoka, Japan) and maintained at a constant temperature of 22 ± 2 °C and a 12 h light–dark cycle with free access to water and food.
ReagentUnless otherwise stated, all reagents were purchased from Nacalai Tesque (Kyoto, Japan). BTZ was purchased from Wako Pure Chemical Corporation (Osaka, Japan) and dissolved by dimethyl sulfoxide (DMSO) at a concentration of 1 mM, stored at −20 °C for future use.
Primary SC Cultures and DifferentiationPrimary SCs were prepared as described previously, with modifications.14) SC proliferative medium was modified to Advanced Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium (Sigma-Aldrich, St. Louis, MO, U.S.A.) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 1% penicillin-streptomycin, 2 mM L-glutamine, 2 µM forskolin (Sigma-Aldrich), and 20 ng/mL recombinant human heregulin-β (PeproTech, Cranbury, NJ, U.S.A.). Differentiation started when cells reached confluency about 95%, and the differentiation period was prolonged from 2 to 5 d. Undifferentiated cells (immature cells) were harvested immediately before differentiation began. Cells were plated at a density of 4.8 × 104 cells/well onto 12-well plates and 1.6 × 103 cells/glass coverslips.
Primary Macrophage CulturesPrimary macrophages were prepared as described previously, with modifications.15) Macrophages were re-suspended in macrophage-maintaining medium (RPMI 1640 medium from Gibco (Carlsbad, CA, U.S.A.), supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), and 1% penicillin–streptomycin), and plated at the density of 1 × 106 cells per well onto 48-well plates. Cells were treated within 2 d after isolation and the treatment medium was shifted to serum-free medium (Advanced DMEM/F12 (Sigma-Aldrich), supplemented with 1% penicillin-streptomycin and 2 mM of L-glutamine).
Primary DRG Neuron CulturesPrimary DRG neurons were prepared as described previously, with modifications.14) Laminin coating was changed to 3% Matrigel Matrix (Corning, Corning, NY, U.S.A.; diluted in ADMEM/F12). Cells were plated at a density of 1.2 × 105 cells/well in 48-well plates and 1 × 105 cells/glass coverslip and treated within 48 h after isolation.
Primary SGC CulturesPrimary SGCs were prepared from purified DRG neurons with removal of neurofilaments to prevent contamination of SCs and other similar cells. DRG neurons were maintained in SC proliferative medium for another 3 d. After reaching over 80% confluency, proliferated SGCs were digested with 0.25% trypsin (Nacalai Tesque). Cells were collected and laid onto a 30% Percoll (Sigma-Aldrich)-70% L-15 medium (GE Healthcare, Grand Island, NY, U.S.A.) cushion for isodensity centrifugation at 1400 × g for 15 min. Only the upper layer of cells was collected (purified SGCs). SGCs were plated onto a surface coated with 0.01% poly-L-lysine (Sigma-Aldrich) at a density of 1.2 × 104 cells/well in 48-well plates and 1.6 × 103 cells/glass coverslip. During the drug treatment, cells were transferred to serum-free medium.
Immunofluorescence Staining and QuantificationAll cell types in this study were plated onto 10 mm coverslips and cultured for drug treatment. Coverslips were fixed with 4% paraformaldehyde aqueous solution for 30 min at room temperature and then rinsed with phosphate-buffered saline (PBS) three times. For the blocking step, 3% albumin bovine serum solution (buffered by 0.1% Triton X-100 supplemented PBS) was applied to each sample and incubated for 30 min at room temperature. The samples were then rinsed five times with PBS and incubated with 60 µL of the following diluted primary antibodies overnight at 4 °C: rat anti-myelin basic protein (MBP, Cat#MAB386, 1 : 500, Merck, Darmstadt, Germany), mouse anti-nerve growth factor receptor (NGFR, P75NTR, Cat#MAB365, 1 : 500, Merck), rabbit anti-glial fibrillary acidic protein (GFAP, Cat#ab7260, 1 : 1000, Abcam, Cambridge, U.K.), mouse anti-β-III tubulin (TUBB3, Cat#801213, 1 : 500, BioLegend, San Diego, CA, U.S.A.) and rabbit anti ionized calcium-binding adapter molecule 1 (Iba1, Cat# 019-19741, 1 : 500, Wako Pure Chemical Corporation). The samples were again rinsed five times with PBS and then incubated with the following diluted secondary antibodies at room temperature for another 2 h: donkey anti-rat immunoglobulin G (IgG) conjugated to Alexa Fluor™ 488 (Cat#A21208, 1 : 200, Thermo Fisher Scientific, Waltham, MA, U.S.A.) for MBP, donkey anti-mouse IgG conjugated to Alexa Fluor™ 594 (Cat#A21203, 1 : 200, Thermo Fisher Scientific) for P75NTR or TUBB3, donkey anti-rabbit IgG conjugated to Alexa Fluor™ 594 (Cat#A21207, 1 : 200, Thermo Fisher Scientific) for GFAP and donkey anti-rabbit IgG conjugated to Alexa Fluor™ 488 (Cat#A21206, 1 : 200, Thermo Fisher Scientific) for GFAP or Iba1. The samples were then rinsed five times with PBS, after which 6 µL of Fluoromount-G (SouthernBiotech, Birmingham, AL, U.S.A.) was applied, and the samples were sealed with nail polish.
Images were captured by the Fluoview FV10i system (Olympus, Tokyo, Japan). For SCs and SGCs, four independent samples with 1–3 fields each were randomly selected from each group. For DRG neurons, four independent samples with 3 fields each were randomly selected from each group. The fluorescence intensity of SCs and SGCs, the covering area of neurite per cell, and degeneration index in DRG neurons were all analyzed with ImageJ software (National Institute of Mental Health, Bethesda, MD, U.S.A.). The average MBP, P75NTR, and GFAP fluorescence intensity per cell was calculated by subtracting background fluorescence intensity from total fluorescence intensity; the result was divided by cell counts (4′-6-diamidino-2-phenylindole (DAPI)-labeled particles). Neurite-covering areas were calculated from images of TUBB3-stained neurons that were processed by the function “Binary”; positive pixels were recognized as neurites.14) Calculation of the average coverage of each cell required the neurite-covering areas to be further divided by cell counts (DAPI-labeled particles). The degeneration index corresponds to the relative area of fragmented neurites.12) The area of fragmented neurites was calculated by summing all the particles in images of TUBB3-stained DRG neurons after the “Binary” process. The ratio of fragmented areas versus total neurite-covering areas was the “degeneration index.”
Quantitative RT-PCR (RT-qPCR)RNA extraction and real-time RT-qPCR was performed as previously described.16) Total RNA was extracted with NucleoSpin RNA (TaKaRa Bio, Shiga, Japan), and ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan) was used to establish cDNA libraries from mRNA. Real-time qPCR was performed on the StepOne real-time PCR system (Life Technologies, Carlsbad, CA, U.S.A.) with protocols from THUNDERBIRD SYBR qPCR Mix (TOYOBO). The sequences of primers used are as follows (written as gene name: 5′-forward-3′, 5′-reverse-3′): rat Mbp: GCCTGTCCCTCAGCAG, GTCGTAGGCCCCCTTG; rat P75NTR: CTGCTGCTGCTGCTGATTC, TGCAGGCTTTGCAGCA; rat Krox20: TCTACCCGGTGGAAGA, TGATCATGCCATCTCC; rat Trpa1: CCGCATAGAGCTCCTCAACC, CTGAAGCCATAGGCACACCA; rat Trpv1: AGGGAGATCCATGAACCCGA, AGGTGTCAATGCAGGACAGG; rat Cacna1h: CCTGGAGGAGAGCAACAAGG, GGCTTTCCTGTGCTGTAGGT; rat Cacna2d1: CACGTTTTACACTGTGCCCC, TGACCGGCTCCTGAGATTTG; rat Scn8a: CCAGAAGAACGGGAACGGAA, ACGGTCAGGTTTGGGTTGTT; rat Scn9a: GAACCCATCACGACCACACT, GTTGTCTGAGGCGATACCGT; rat Scn10a: TGGCCGTAGACATGGAGAAGA, CTGTAGTGTCGAGTGCCGGG; rat Scn11a: CGTACCAGGAAGACAGGGTGAA, GGAAGTGAAGGGGCGGAA; and rat Gapdh: GTGCCAGCCTCGTCTCATAG, AGAGAAGGCAGCCCTGGTAA. The first step of PCR was set to 95 °C for 10 min and followed by 40 cycles that looped from 95 °C for 15 s to 60 °C for 60 s. Gapdh was selected as the reference gene for relative quantity analysis, and each gene result was normalized to the Gapdh RNA level.
Measurement of Secreted Protein in Culture MediumConcentrations of tumor necrosis factor-α (TNFα), interleukin-1 beta (IL1β), C-C Motif Chemokine Ligand 5 (CCL5), interleukin-6 (IL-6), C-X-C motif chemokine ligand 1 (CXCL1), and C-X-C motif chemokine ligand 2 (CXCL2) in the culture medium of either SGCs or macrophages were measured by matched enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, U.S.A.), following the manufacturer’s instructions.
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium Bromide (MTT) AssayFor the assay, 0.1% MTT (Nacalai Tesque) in PBS was added to the culture medium at a ratio of 1 : 5 and incubated with cells at 37 °C with 5% CO2 for 30 min. After removing the medium, the resultant formazan crystals were dissolved in 200 µL DMSO and transferred into a clear-bottomed 96-well plate. Sample absorbance was measured at 570 nm with an ELISA analyzer (Bio-Rad, Hercules, CA, U.S.A.). Cell viability was expressed as a ratio in comparison with vehicle groups.
Statistical AnalysisAll data were expressed as mean ± standard error of the mean (S.E.M.). Statistical analysis was performed by the Student’s t-test using GraphPad Prism 9 (GraphPad Software, San Diego, CA, U.S.A.). p < 0.05 was considered statistically significant.
RESULTS
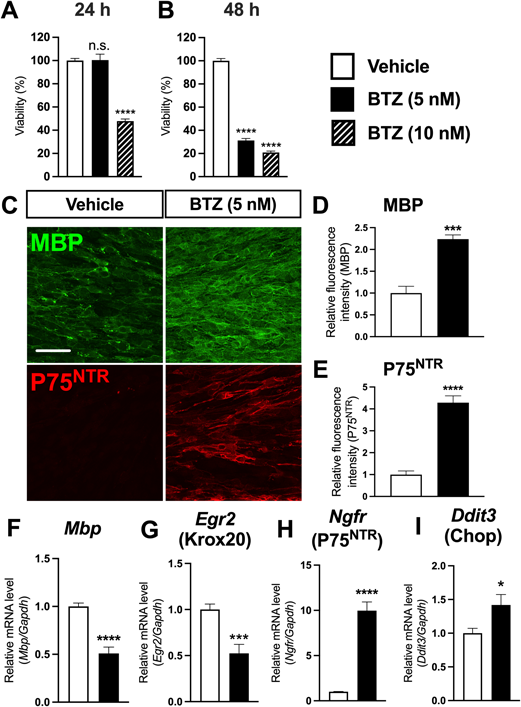
BTZ Directly Induces Dedifferentiation of Mature SCsWe first confirmed the differentiation and dedifferentiation of a cultured SC experimental system. SCs from the sciatic nerves of neonatal rats were used to evaluate the direct effects of BTZ. SCs in proliferative medium are recognized as immature. To obtain mature SCs, cells were transferred to serum-free medium supplemented with a higher forskolin concentration (20 µM) for prolonged differentiation (5 d). Mature SCs were characterized by a significant drop in the expression level of P75NTR (Supplementary Fig. 1A), a marker of immature SCs, and a significant increase in the expression levels of Mbp (Supplementary Fig. 1B) and Krox20 (Supplementary Fig. 1C), which are generally considered to be credible markers of mature SCs. Moreover, these results were further supported by the P75NTR and MBP protein levels, quantified as fluorescence intensity, in mature and immature SCs (Supplementary Figs. 1D, E). Images of both MBP and P75NTR in mature and immature SCs are also shown (Supplementary Fig. 1F).
Because the differentiation process of SCs has been firmly validated, BTZ treatments were performed on this culture system. Relatively high concentrations (24 h, 10 nM) and relatively long term (48 h, 5 nM) of BTZ significantly reduced SCs viability, while BTZ at 5 nM for 24 h did not affect SCs viability (Figs. 1A, B). Therefore, BTZ was applied to cells in this study for 24 h and 5 nM, which is similar to that in clinical use.17) Administration of BTZ at 5 nM for 24 h increased both MBP and P75NTR protein levels in mature SCs (Figs. 1C–E). BTZ is known for its role in inducing abnormal protein accumulation,6) and thus further assessing changes in RNA levels could better define the onset of dedifferentiation. Thus, MBP and P75NTR RNA levels were examined under the same conditions. In BTZ-treated mature SCs, Mbp (Fig. 1F) and Egr2 (Krox20, Fig. 1G) mRNA levels were decreased, whereas mRNA level of Ngfr (P75NTR, Fig. 1H) were greatly increased. To confirm BTZ-induced proteasome inhibition, we examined Ddit3, an endoplasmic reticulum stress marker, and found that mRNA expression level of Dtit3 was also increased. These results clearly demonstrate that the RNA level changes in myelination biomarkers in BTZ-treated mature SCs were similar to those in immature SCs, implying that BTZ reverses SC maturation. MBP protein accumulation can most likely be interpreted as the result of protein degeneration failure caused by BTZ cytotoxicity rather than increased de novo biosynthesis.
BTZ Upregulates GFAP Expression in SGCs without Causing InflammationSGCs derived from primary DRG neurons of rats were used to evaluate the direct effects of BTZ. The purity of the primary cultures was validated by immunostaining GFAP expressed in SGCs (Supplementary Figs. 2A, B). In rodent in vivo models, SGC activation is a typical feature of DRG local inflammation, which is generally and exclusively observed as GFAP upregulation.6) However, it is unresolved whether GFAP upregulation is directly related to the release of pro-inflammatory cytokines in BTZ models.6) BTZ increased GFAP protein levels in cultured SGCs (Figs. 2A, B). However, this outcome did not result from upregulation of GFAP mRNA expression (Fig. 2C). Furthermore, the secreted cellular factors analyzed by ELISA did not indicate any sign of inflammation; BTZ-treated SGCs failed to release cytokines, e.g., TNFα (Fig. 2D) and IL1β (Fig. 2E), or chemokines, e.g., CCL5 (Fig. 2F). These results suggest that BTZ-induced GFAP upregulation in cultured SGCs can be a direct result of BTZ-induced protein accumulation rather than strong inflammation. This observation most likely provides an alternative explanation for the upregulated GFAP observed at the DRG site in BTZ in vivo models. Additionally, cell viability was not altered during these BTZ-induced pathological changes (Fig. 2G).
BTZ Exhibits Mild Cytotoxicity but Hardly Induces Inflammatory Reactions in MacrophagesNeuroinflammation has been reported to occur in mouse models of BIPN.6) Therefore, we examined the effects of BTZ on macrophages, immune cells responsible for peripheral inflammation. Culture purity of mouse peritoneal macrophages was validated by immunostaining Iba1 (Supplementary Fig. 3). Unexpectedly, BTZ did not affect the expression levels of the inflammatory biomarkers tested (Figs. 3A–E) or the viability of macrophages (Fig. 3F).
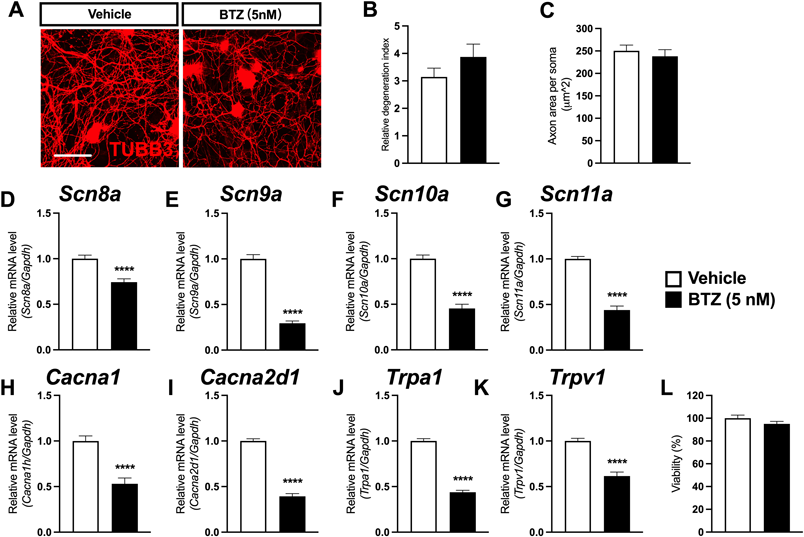
BTZ Does Not Alter Neurite Outgrowth or Integrity but Affects the Expression Levels of Sensor and Transducer Ion ChannelsDRG neurons were collected from thoracic and lumbar DRGs of rats, and culture purity was validated by immunostaining TUBB3 (Supplementary Fig. 2C). BTZ hardly caused any detectable changes in DRG neuron morphology (Fig. 4A) and exerted no significant effects on neuron toxicity, characterized by the fragmentization rate (Fig. 4B) and neurite density (Fig. 4C). Next, the expression levels of transducer or sensor ion channels, ion channels (Scn8a, Scn9a, Scn10a, Scn11a, Cacna1h, Cacna2d1, and Trpa1, Trpv1), and voltage-gated channel alpha subunits (Navs) were evaluated, which are involved in BIPN development in DRG neurons.18) BTZ significantly reduced Scn8a (Nav1.6; Fig. 4D), Scn9a (Nav1.7; Fig. 4E), Scn10a (Nav1.8; Fig. 4F), and Scn11a (Nav1.9; Fig. 4G) expression levels. Similarly, the expression levels of the calcium voltage-gated channel subunits Cacna1h (Cav3.2) and Cacna2d1 (Cavα2δ1) (Figs. 4H, I) were both downregulated by BTZ.
The only two sensor ion channels included in this examination, Trpa1 (TRPA1; Fig. 4J) and Trpv1 (TRPV1; Fig. 4K), were also downregulated by BTZ. However, cell viability was not altered when these changes occurred (Fig. 4L). Although the channels included in this study exhibit much diversity, the alterations to them can generally be recognized as a firm sign of DRG neuron dysfunction that is detectable even before changes to cell morphology or viability.
DISCUSSION
In this study, we examined the effects of 5 nM BTZ on four different cell cultures and found that BTZ dedifferentiated SCs, increased GFAP protein levels in SGCs without inducing inflammation, and reduced ion channel expression in DRG neurons, whereas it did not stimulate macrophages.
Previous in vitro reports preferred to apply harsh conditions to DRG neurons (i.e., > 50 nM BTZ for 24 h)11,19) and milder conditions to SCs (i.e., approx. 50 nM BTZ for up to 18 h).11,20) According to previous findings on BTZ pharmacokinetics, the blood concentration of BTZ reaches Cmax at around 200 nM within 30 min after injection.17) In that regard, a concentration of 50–100 nM would suffice for analyzing acutely elicited adverse effects; however, the concentration then drops sharply to ≤5 nM within 3 h.17) Therefore, concentrations as high as 50–100 nM are not appropriate for studying the chronic adverse effects of BTZ, whereas concentrations as low as 5 nM (used in this study) are considered appropriate. Additionally, BTZ is considered to freely across the cell membrane according to U.S. Food and Drug Administration (FDA) documents21) and also reported that the intracellular concentration of BTZ is not related to variable transporter expression in myeloma cells.22) This indicates that the uptake or efflux of BTZ would be simplified to physical action without tissue-specified enrichment. Thus, a series of validation tests on cell viability were performed, and 5 nM BTZ for 24 h was selected as a universal treating condition for all cell types in this study. BTZ did not affect the viability of any cell type but still strong enough to cause changes at the molecular level.
In BTZ-treated SCs, the aggregation of misfolded protein is considered to trigger cellular metabolic disorder and secondarily induce endoplasmic reticulum stress.11,20) which is associated with our data showing that BTZ-induced increase in mRNA expression of Dtim3, an endoplasmic reticulum stress marker. In an earlier report, cultured primary SCs that treated with a high dose of BTZ exhibited decreased numbers of myelin-related proteins.11) Our current study is the first to demonstrate that BTZ (at low concentrations) can induce the dedifferentiation of SCs (as observed by increased levels of markers of immature SCs), thus possibly causing reversible demyelination in vivo. Demyelination in BTZ-treated rodent models has been firmly validated, whereas the irretrievable loss of SCs or only temporary dysfunction of myelination have yet to be demonstrated.10,11,20) Our current study provides an alternative possibility to explore. In present study, BTZ increased MBP levels but decreased Mbp RNA levels in SCs, implying that abnormally accumulated myelin protein may become an autoimmune antigen that recruits immune cells to infiltrate, which has been reported in an in vivo model.11,20)
SGCs are known for their close developmental relationship with SCs but differ from SCs in that they are not myelin-forming cells and retain their plasticity throughout their lifespan.23) Our study provides the first viable model of BTZ-induced effects on SGCs in vitro. GFAP upregulation was clearly observed in BTZ-treated SGCs and is unlikely to be the result of upregulated biosynthesis but rather unexpected protein accumulation (in this model). It has been believed that increasing GFAP levels in glial cells are closely related to inflammatory glial activation in BIPN models; however, this assertion is undermined by the above-mentioned result.6) Because it is possible that solely GFAP is increased by BTZ, it is necessary to understand the effect of GFAP overexpression in glial cells. A recent report revealed that GFAP overexpression in astrocytes in vitro exerts myelin-protective effects and inhibits the release of inflammatory chemokines (e.g., CCL2 and CXCL10). This could provide an alternative explanation: SGCs play an immunosuppressive role against the effects of BTZ.24) Taken together, the finding that BTZ-induced GFAP upregulation is independent of inflammation contradicts the previous notion that exclusively neuronal inflammation causes increased GFAP levels at DRG sites. However, additional data are needed to clarify the function of SGCs in the development of BIPN.
Macrophages are immunoreactive by polarizing to a pro-inflammatory (M1) or an anti-inflammatory (M2) state under stimulation. BTZ-induced macrophage infiltration has been confirmed, and most researchers consider its appearance a sign of neuronal inflammation.6,25) This conclusion contradicts the results of our current study and previous research on the autoimmune-suppressive role of BTZ.26) A possible explanation for this inconsistency is that the inflammation that occurs at local DRGs is an indirect consequence of BTZ treatment, and secondary to the damaged neuronal somas or DRG glial cells caused by BTZ, while stress or apoptosis signals possibly trigger the recruitment of macrophages or T-cells. In our study, BTZ did not induce macrophages to release pro-inflammatory cytokines, which is in agreement with the clinical outcomes of BTZ. Strictly speaking, only IL1β showed a slight tendency of decrease with no significance (p = 0.0823, Fig. 3B). This is reminiscent of caspase-1-mediated proteolytic cleavage for IL1β production pathway, unlike other pro-inflammatory cytokines including TNFα and IL6. Recent study reported that precursor IL1β is rapidly turned over by the proteasome and this correlates with its decoration by ubiquitin chains,27) implying that proteasome inhibition by BTZ may affect the ubiquitylation of IL1β and resultant production of bioactive IL1β protein. Further multifaceted experiments will be needed to firmly validate this observed effect. In any case, its role in BIPN development is unclear. Nevertheless, this study isolated mouse peritoneal macrophages for BTZ treatment, whereas in both human and mouse DRGs, there is an unneglectable number of tissue-resident macrophages, which may differ from peritoneal macrophages under the same drug stimulation.28) Further investigations are needed to determine how BTZ affects tissue-resident macrophages and other non-glial cells.
After the termination of BTZ treatment, clinical symptoms usually return, and BTZ only affects motor functions at a minor level.29) Furthermore, the decisive anatomical difference between motor and sensory nerves is considered to be the position of neuronal soma. Taken together, harmful exposure to anti-cancer agents only affects DRG neurons and not motor neurons, which is possibly essential in neuropathy development. In previous research, BTZ at high concentrations exerted strong effects, and neurites displayed a strong trend toward fragmentization. The downstream mechanism of BTZ-induced neurite destruction has only recently been revealed to be activation of SARM1, a direct executor of multi-origin Wallerian degeneration.6,30) Another study also highlighted that exposure of neuronal somas to BTZ was necessary for axon degeneration, supporting the hypothesis that soma exposure is necessary in vivo.19) However, events downstream of the SARM1 pathway remain elusive.
Furthermore, BTZ-induced damage to mitochondria trafficking in DRG neurons and somatic TUBB3 aggregation has been reported.31) Only recently has the underlying mechanism been revealed to be an increase in δ-II tubulin, the result of a hyper-stable tubulin posttranslational modification, which was also abnormally detected in the DRG neurons of BIPN patients. This was sufficient to cause both axon destruction and mitochondria stagnation in cultured DRG neurons.12)
However, nearly all the mentioned studies on cultured DRG neurons were based on harsh conditions of BTZ treatment (> 100 nM and usually over 24 h). These conditions greatly deviate from clinical observations, and thus the Wallerian degenerations reported by those in vitro studies cannot represent actual clinical axonopathy because no fragmentization or permanent axon loss was reported. The model developed in our current study has demonstrated that, even under a much milder condition (under which neurites are hardly damaged), the expression levels of ion channels related to sensing are already strongly disrupted, which can account for both BIPN development and clinical observations. In cultured DRG neurons, BTZ exerted notable effects by downregulating most pain-related ion channels. In relation to BIPN, numerous reports have observed upregulation of TRPA1, TRPV1, and Cav3.2 in DRG neurons in in vivo rodent models32–35); However, no studies have reported a direct influence of BIPN on sensory ion channels located on cultured DRG neurons, and his makes it difficult to distinguish between the direct and indirect effects of BTZ treatment. Most patients not only develop hypersensitivity or pain, which has already been widely validated by rodent models, but also the stocking-glove pattern of symptoms.36) These symptoms have hardly received any attention because rodent behaviors are usually mixed with painful symptoms, making any clear identification impossible. Sciatic nerve signal transmission has been reported to be damaged during BTZ treatment, possibly owing to myelin sheath loss; however, this inference was undermined by the fact that motor nerves were not affected.10,11) Furthermore, the reduced excitability of neurons in BIPN has been supposed to be caused by mainly soma neurotoxicity.19) Here, we have provided molecular evidence of ion channels for this hypothesis for the first time. The homeostasis of ion channels (which modify and shape signals from afferent nerves in the sensory system) is disrupted, and abnormalities in their biosynthesis may result in overall signal transmission failure.
Acknowledgments
We would like to thank Ms. Mari Suzuki for her very helpful advice on all aspects of this study. We would also like to thank Dr. Soni Siswanto for primary macrophage cultures, Dr. Hiroyuki Kawai for primary SGC and DRG cultures, and Mr. Qiubin Yang for primary SC cultures. This work was supported by Grants-in-Aid for Scientific Research (KAKENHI) from MEXT/JSPS (to H.S., JP19H03377, and to S.K., JP20H00491) and by the Nakatomi Foundation and Takeda Science Foundation (to H.S.).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Besse A, Besse L, Kraus M, Mendez-Lopez M, Bader J, Xin BT, de Bruin G, Maurits E, Overkleeft HS, Driessen C. Proteasome inhibition in multiple myeloma: head-to-head comparison of currently available proteasome inhibitors. Cell Chemical Biology, 26, 340–351.e3 (2019).
- 2) Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets, 11, 239–253 (2011).
- 3) Lee SE, Choi K, Han S, Lee J, Hong T, Park GJ, Yim DS, Min CK. Bortezomib pharmacokinetics in tumor response and peripheral neuropathy in multiple myeloma patients receiving bortezomib-containing therapy. Anticancer Drugs, 28, 660–668 (2017).
- 4) Albornoz N, Bustamante H, Soza A, Burgos P. Cellular responses to proteasome inhibition: molecular mechanisms and beyond. Int. J. Mol. Sci., 20, 3379 (2019).
- 5) Ezendam NP, Pijlman B, Bhugwandass C, Pruijt JF, Mols F, Vos MC, Pijnenborg JM, van de Poll-Franse LV. Impact of chemotherapy-induced neurotoxicities on adult cancer survivors’ symptom burden and quality of life. J. Cancer Surviv., 12, 234–245 (2018).
- 6) Moschetti G, Amodeo G, Maftei D, Lattanzi R, Procacci P, Sartori P, Balboni G, Onnis V, Conte V, Panerai A, Sacerdote P, Franchi S. Targeting prokineticin system counteracts hypersensitivity, neuroinflammation, and tissue damage in a mouse model of bortezomib-induced peripheral neuropathy. J. Neuroinflammation, 16, 89 (2019).
- 7) Foran E, Kwon DY, Nofziger JH, Arnold ES, Hall MD, Fischbeck KH, Burnett BG. CNS uptake of bortezomib is enhanced by P-glycoprotein inhibition: implications for spinal muscular atrophy. Neurobiol. Dis., 88, 118–124 (2016).
- 8) Huehnchen P, Springer A, Kern J, Kopp U, Kohler S, Alexander T, Hiepe F, Meisel A, Boehmerle W, Endres M. Bortezomib at therapeutic doses poorly passes the blood–brain barrier and does not impair cognition. Brain Communications, 2, fcaa021 (2020).
- 9) Robinson CR, Dougherty PM. Spinal astrocyte gap junction and glutamate transporter expression contributes to a rat model of bortezomib-induced peripheral neuropathy. Neuroscience, 285, 1–10 (2015).
- 10) Cavaletti G, Gilardini A, Canta A, Rigamonti L, Rodriguez-Menendez V, Ceresa C, Marmiroli P, Bossi M, Oggioni N, D’Incalci M, De Coster R. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp. Neurol., 204, 317–325 (2007).
- 11) Shin YK, Jang SY, Lee HK, Jung J, Suh DJ, Seo SY, Park HT. Pathological adaptive responses of Schwann cells to endoplasmic reticulum stress in bortezomib-induced peripheral neuropathy. Glia, 58, 1961–1976 (2010).
- 12) Pero ME, Meregalli C, Qu X, Shin GJ, Kumar A, Shorey M, Rolls MM, Tanji K, Brannagan TH, Alberti P, Fumagalli G, Monza L, Grueber WB, Cavaletti G, Bartolini F. Pathogenic role of delta 2 tubulin in bortezomib-induced peripheral neuropathy. Proc. Natl. Acad. Sci. U.S.A., 118, e2012685118 (2021).
- 13) Argyriou AA, Cavaletti G, Bruna J, Kyritsis AP, Kalofonos HP. Bortezomib-induced peripheral neurotoxicity: an update. Arch. Toxicol., 88, 1669–1679 (2014).
- 14) Imai S, Koyanagi M, Azimi Z, Nakazato Y, Matsumoto M, Ogihara T, Yonezawa A, Omura T, Nakagawa S, Wakatsuki S, Araki T, Kaneko S, Nakagawa T, Matsubara K. Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Sci. Rep., 7, 5947 (2017).
- 15) Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S. TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J. Neurosci., 32, 3931–3941 (2012).
- 16) Ohashi K, Shibasaki K, Nakazawa H, Kunimasa R, Nagayasu K, Shirakawa H, Kaneko S. Transient receptor potential melastatin 3 is functionally expressed in oligodendrocyte precursor cells and is upregulated in ischemic demyelinated lesions. Biol. Pharm. Bull., 44, 181–187 (2021).
- 17) Moreau P, Karamanesht II, Domnikova N, Kyselyova MY, Vilchevska KV, Doronin VA, Schmidt A, Hulin C, Leleu X, Esseltine DL, Venkatakrishnan K, Skee D, Feng H, Girgis S, Cakana A, van de Velde H, Deraedt W, Facon T. Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin. Pharmacokinet., 51, 823–829 (2012).
- 18) Aromolaran KA, Goldstein PA. Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy. Mol. Pain, 13, 1744806917714693 (2017).
- 19) Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, DiAntonio A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight, 4, e129920 (2019).
- 20) Watanabe T, Nagase K, Chosa M, Tobinai K. Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br. J. Cancer, 103, 1580–1587 (2010).
- 21) Center of Drug Evaluation and Research, FDA. “21602 Velcade Pharmacology review part 1 (p. 8).”: ‹http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_pharmr_P1.pdf›, accessed 3 November, 2022.
- 22) Clemens J, Seckinger A, Hose D, Theile D, Longo M, Haefeli WE, Burhenne J, Weiss J. Cellular uptake kinetics of bortezomib in relation to efficacy in myeloma cells and the influence of drug transporters. Cancer Chemother. Pharmacol., 75, 281–291 (2015).
- 23) George D, Ahrens P, Lambert S. Satellite glial cells represent a population of developmentally arrested Schwann cells. Glia, 66, 1496–1506 (2018).
- 24) Kramann N, Menken L, Pförtner R, Schmid SN, Stadelmann C, Wegner C, Brück W. Glial fibrillary acidic protein expression alters astrocytic chemokine release and protects mice from cuprizone-induced demyelination. Glia, 67, 1308–1319 (2019).
- 25) Meregalli C, Marjanovic I, Scali C, Monza L, Spinoni N, Galliani C, Brivio R, Chiorazzi A, Ballarini E, Rodriguez-Menendez V, Carozzi VA, Alberti P, Fumagalli G, Pozzi E, Canta A, Quartu M, Briani C, Oggioni N, Marmiroli P, Cavaletti G. High-dose intravenous immunoglobulins reduce nerve macrophage infiltration and the severity of bortezomib-induced peripheral neurotoxicity in rats. J. Neuroinflammation, 15, 232 (2018).
- 26) Verbrugge SE, Scheper RJ, Lems WF, de Gruijl TD, Jansen G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther., 17, 17 (2015).
- 27) Vijayaraj SL, Feltham R, Rashidi M, Frank D, Liu Z, Simpson DS, Ebert G, Vince A, Herold MJ, Kueh A, Pearson JS, Dagley LF, Murphy JM, Webb AI, Lawlor KE, Vince JE. The ubiquitylation of IL-1β limits its cleavage by caspase-1 and targets it for proteasomal degradation. Nat. Commun., 12, 2713 (2021).
- 28) Avraham O, Chamessian A, Feng R, Yang L, Halevi AE, Moore AM, Gereau RW 4th, Cavalli V. Profiling the molecular signature of satellite glial cells at the single cell level reveals high similarities between rodents and humans. Pain (2022), in press.
- 29) Yamamoto S, Egashira N. Pathological mechanisms of bortezomib-induced peripheral neuropathy. Int. J. Mol. Sci., 22, 888 (2021).
- 30) Staff NP, Podratz JL, Grassner L, Bader M, Paz J, Knight AM, Loprinzi CL, Trushina E, Windebank AJ. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology, 39, 124–131 (2013).
- 31) Guo L, Hamre J 3rd, Eldridge S, Behrsing HP, Cutuli FM, Mussio J, Davis M. Multiparametric image analysis of rat dorsal root ganglion cultures to evaluate peripheral neuropathy-inducing chemotherapeutics. Toxicol. Sci., 156, 275–288 (2017).
- 32) Liu D, Sun M, Xu D, Ma X, Gao D, Yu H. Inhibition of TRPA1 and IL-6 signal alleviates neuropathic pain following chemotherapeutic bortezomib. Physiol. Res., 68, 845–855 (2019).
- 33) Li C, Deng T, Shang Z, Wang D, Xiao Y. Blocking TRPA1 and TNF-α signal improves bortezomib-induced neuropathic pain. Cell. Physiol. Biochem., 51, 2098–2110 (2018).
- 34) Quartu M, Carozzi VA, Dorsey SG, Serra MP, Poddighe L, Picci C, Boi M, Melis T, Del Fiacco M, Meregalli C, Chiorazzi A, Renn CL, Cavaletti G, Marmiroli P. Bortezomib treatment produces nocifensive behavior and changes in the expression of TRPV1, CGRP, and substance P in the rat DRG, spinal cord, and sciatic nerve. Biomed. Res. Int., 2014, 180428 (2014).
- 35) Tomita S, Sekiguchi F, Deguchi T, Miyazaki T, Ikeda Y, Tsubota M, Yoshida S, Nguyen HD, Okada T, Toyooka N, Kawabata A. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology, 413, 33–39 (2019).
- 36) Chaudhry V, Cornblath DR, Polydefkis M, Ferguson A, Borrello I. Characteristics of bortezomib- and thalidomide-induced peripheral neuropathy. J. Peripher. Nerv. Syst., 13, 275–282 (2008).