Review
Innovative System for Delivering Nucleic Acids/Genes Based on Controlled Intracellular Trafficking as Well as Controlled Biodistribution for Nanomedicines
2023 年 46 巻 12 号 p. 1648-1660
詳細
2023 年 46 巻 12 号 p. 1648-1660
This review paper summarizes progress that has been made in the new field of “Controlled Intracellular Trafficking.” This involves the development of new systems for delivering plasmid DNA (pDNA), small interfering RNA (siRNA), mRNA, proteins, their escape from endosomes, the mechanism for how they enter the nucleus, how they enter mithochondria and how materials subsequently function within a cell. In addition, strategies for delivering these materials to a selective tissue after intravenous administration was also intensively investigated not only to the liver but also to tumors, lungs, adipose tissue and the spleen. In 2020, a new mRNA vaccine was developed against coronavirus disease 2019 (COVID-19), where ionizable cationic lipids were used as a delivery system. Our strategy to identify an efficient ionizable cationic lipids (iCL) based on a lipid library as well as their applications concerning the delivery of siRNA/mRNA/pDNA is also described.
Pharmacokinetics (PK) was my favorite subject in my School of Pharmaceutical Sciences, especially physiologically based pharmacokinetics (PBPK) which encouraged me to choose the laboratory where I was engaged in constructing a PBPK model of quinidine, as a model compound of weak basic drugs in rats. I was measuring the time courses for quinidine levels in plasma, liver, lung, heart, kidney, brain, gastrointestinal (GI), muscle, adipose and skin for estimating the tissue to plasma concentration ratio as well as the unbound fraction in plasma and blood to plasma concentration ratio to construct a PBPK model of quinidine. The hepatic intrinsic clearance of quinidine was estimated from the time course for the plasma concentration of quinidine. I found that constructing a PBPK model for quinidine was exciting but, since then, many PBPK models have been developed in our laboratory for compounds such as ethoxybenzamide, tolbutamide, diazepam, doxorubicin. I recognized that there was little originality for my PBPK model of quinidine, it was simply another example. My supervisor Professor Sugiyama found that the Kp-values of quinidine were non-linear in most tissues and this finding was incorporated into my PBPK model and was published.1) The main subject in our laboratory at that time was “Prediction of PK: from in vitro to in vivo, from normal state to disease state,” which, at that time seemed to be a high impact subject which I found to be exciting. The next subject I chose for my Ph.D. thesis was PK/pharmacodynamics (PD)-modeling and ouabain, which produces a delayed pharmacological effect, was chosen as a model compound. I had to establish a quantitative measure of a pharmacological effect such as positive inotropic action, which occupied my research activities for almost 1 year. It seemed to me that it was a waste of time and energy to establish a pharmacological effect of a certain drug by myself, since I was not a pharmacologist but an expert in the area of PK. I focused on the infusion rate dependent inotropic action of ouabain in rabbits by measuring both the plasma concentration of ouabains and pharmacological effects with different infusion rates.2) A clear infusion rate dependency was observed and the time delay in the appearance of pharmacological effects could be explained by the slow binding/dissociation process of ouabain to/from Na+, K + -ATPase, a receptor of ouabain in the isolated perfused rabbit heart.3) It was also found that the binding of ouabain to Na+, K + -ATPase can be a determining factor in the tissue distribution of ouabain, since there was little non-specific binding of ouabain.4)
After postdoctoral training at Stanford University, I returned to Japan and joined the laboratory of Professor Hiroshi Kiwada who is an expert in the field of liposome research at Tokushima University as an associate professor. It was a good opportunity for me to jump into a new research field of drug delivery system (DDS). He was interested in analyzing interactions between liposomes and serum and he found that the complement system as an important factor in recognizing liposomes.5) I was looking for a new area of DDS for my future research and I was reading a text book of Molecular Cell Biology. When I came across a section on endocytosis, I was inspired to imagine that it should be fantastic if I could control the intracellular trafficking of vesicular transport after endocytosis using liposomes as a tool. I was deeply influenced by the research of Stahl et al. who are analyzing intracellular fate of endosomes such as the maturation or vesicular transport. My interests led me to the issue of how to control the intracellular trafficking of endocytosed materials that were encapsulated into liposomes. After becoming acquainted with the field, I designed a model experiment to deliver albumin into nuclei of macrophages using liposomes as a carrier. Albumin cannot penetrate the nuclear membrane by itself, however, NLS conjugated albumin can be transported into the nucleus by recognizing importin-a. In these experiments, pH-sensitive liposomes were used to deliver albumin from endosomes to the cytosol after endocytosis by macrophages. To measure the nuclear delivery of albumin to the nucleus, albumin was conjugated with fluorescent probe as well as NLS so that they could be recognized by confocal laser scanning microscopy, which was just introduced in Tokushima University. In collaboration with Professor Futaki, albumin conjugated with NLS and a fluorescent probe was encapsulated into pH-sensitive liposomes and was applied to macrophages.6) After careful staining of the nucleus, clear colocalization between fluorescent probe and the nucleus was observed. It was the first experience for me to collaborate with a chemist to confirm a new hypothesis and this successful collaboration increased the power of our future research. I was planning to extend this study from albumin to plasmid DNA (pDNA) for gene therapy. In 1999, an elegant study was reported to enhance the transfection activity of pDNA by introducing NLS at the edge of linearized pDNA by Behr and colleagues.7) The basic concept was the same as ours for delivering albumin to the nucleus, however, their design was very good and we became convinced that their findings confirmed the superiority of their concept. Since our attempts regarding gene therapy was blocked by this elegant study, I changed my research direction to analytical aspects, since I was interested in being as productive as I could in order to be eventually promoted to the rank of professor. I had a simple question, namely, how many pDNAs would need to be delivered to the nucleus to produce a significant level of proteins coded by a pDNA. We considered that PCR methodology may be the best for this analysis, since it is sensitive and quantitative enough to permit the number of pDNAs in the nucleus to be evaluated. Since I had no experience in the use of pDNA in my studies at that time, I collaborated with Professor Yasuo Shinohara who is a molecular biologist to pursue this research. It was not so hard to establish a PCR-method to quantify the numbers of pDNA molecules in an isolated nucleus from transfected cells, since Professor Shinohara kindly helped us to solve many problems that we were facing. There was a linear relationship between the number of pDNAs in the nucleus and transfection activity in the case of a lower dose, while the transfection activity was saturated at a higher dose.8) The number of pDNA molecules in the nucleus were approximately 103 pDNA/nucleus, which was much higher than we expected.8) It was difficult to convince a respected researcher like Professor Leaf Huang when we invited him to the annual meeting of Pharmaceutical Society of Japan (PSJ) at Tokushima in 1999. Although our methodology was criticized from various view points, I was convinced that, based on our data, that a large number of pDNA molecules could be delivered to the nucleus and that a significant transfection activity could be achieved.
In 1999, I was appointed as a Professor of Graduate School of Pharmaceutical Sciences, Hokkaido University. My first job was to create a name for our new laboratory (Fig. 1). After careful thinking, I concluded that Laboratory for Molecular Design of Pharmaceutics was the best name for a new laboratory, since I wanted to design DDS at the molecular level as chemists design drugs. The second job was to recruit new faculty members, which seemed to be much more difficult than I expected. It took a few years until three faculty members had been recruited. In 2000, associate professor Hiroyuki Kamiya joined our laboratory and played a principal part in “Intranuclear disposition of pDNA was responsible” as well as his nucleic acid mutation research. He was recommended by professor Eiko Ohtsuka who is famous for her work on nucleic acids and she was his mentor. The first collaborative study was on the effect of NLS on the transfection activity of linearized DNA, which was the study I was interested in at Tokushima University due to the report of JP Behr. I was expecting to expand the concept by introducing not only single NLS but also multiple NLS on linearized DNA. The linearized DNA was prepared by Hiroyuki and the attachment of NLS was done by Akira Matsuda who was an expert in the area of nucleic acid chemistry at our school. Unfortunately, there was no enhancement by NLS on the transfection activity,9) which was very disappointing for us. According to JP Behr, he could not reproduce the data after his colleague left his laboratory. Considering NLS related works on nuclear delivery and transfection activity, enhancing pDNA into the nucleus with NLS was a difficult task, but proteins can be transferred into nucleus efficiently.

In 2002, Dr. Hidetaka Akita joined our laboratory as an assistant professor and played a principal part in the “nuclear delivery of pDNA” as well as “in vivo disposition of DDS.” He was recommended by professor Yuichi Sugiyama who was his supervisor and is also my mentor. The first study was to establish a quantitative analytical method for the intracellular trafficking of pDNA as well as DDS carriers. We were challenged to quantify the intracellular fate of pDNA as well as DDS carriers using confocal laser scanning microscopy (CLSM), since there was no methodology available for quantifying pDNA in endosomes, cytosol, nucleus, etc. It was really hard work to quantify intensities from pDNA which are co-localized with endosomes or the nucleus even in a single z-section within a cell. We checked 30 z-sections from a single cell. In addition, we needed to gather information from 50 cells to account for statistical variations. It was found that 30 cells were sufficient to obtain statistically stable variations. Finally, we succeeded in establishing a quantitative three-dimensional analysis of the intracellular trafficking of pDNA using CLSM.10) We then applied this method to compare the efficiency of each step in intracellular trafficking of DNA after transfection by viral and non-viral vectors. It was known that viral vectors are much more efficient than non-viral vectors in transfection activity, however, there was little information concerning the magnitude of the differences, a step that is important to differentiate such differences. We had an interesting question and a methodology to answer it. This study was the subject for the Ph.D. degree for Susumu Hama. According to his preliminary results, there were nearly 103–104 differences in transfection efficiency between adenoviral vectors and non-viral vectors. In which process could we find such a big difference? After many experiments, it was found that there were no big differences in cellular uptake, endosomal escape and nuclear delivery, however, there was almost a 104 difference after reaching the nucleus (Fig. 2). This result strongly indicated that it was transcriptional/translational processes where the 104 differences were induced. We were surprised to see this result and checked the experimental procedures many times. It was beyond our expectations and we hesitated to submit this amazing result for publication in a few months. Finally, we submitted our manuscript and received a recommendation of revision. We were surprised to see the comments of the reviewer that the authors should repeat the same study in a different cell line, since this result was surprising and difficult for us to carry out. It took almost one year to complete this study and it seemed difficult to complete within 2 months as requested by the reviewer. We decided to repeat the study in a different cell line. It was also surprising that the result in a different cell line was almost the same as the first one. Our revision was accepted at once after submission with confirmation by our repeated experiments.11) We were encouraged to identify the rate limiting step in transfection activity, transcription or translation. By measuring mRNA levels, we were able to divide transcriptional process and translational process. It was concluded that translational process contributed to the difference in transfection activity between viral and non-viral vectors by 460-fold, while a 16-fold difference was found for the transcriptional process.12)

Obtaining research grants is a continuing concern. Core Research for Evolutionary Science and Technology (CREST) is famous for its high standard of research quality as well as the large size of their grants, however, we can only apply for these when the subject of the research field opened fits well with our own research subject. In 2000, a new research field opened for DDS researchers to apply to. When I was wondering how to apply it, Professor Kataoka invited me to apply to CREST for a joint project. After long and intensive collaboration, we were able to prepare successful application form for CREST, which was evaluated highly by the committee. I learned a lot from Professor Kataoka concerning how to make a good application for grants, how to develop a 5 year research plan as well as a 1 year plan. Based on this wonderful collaboration, we were able to accelerate our research as fast as possible and as efficient as possible. The most important factor for me in this collaboration was acquiring Dr. Kentaro Kogure from Tokushima University. He played an essential role in our research team. A number of successful research projects were introduced.
4.1. Introduction of Cell Penetrating Peptide, R8As mentioned above, our collaboration was successful when I asked Professor Futaki to conjugate NLS to albumin for nuclear transport. After promotion to Professor at Kyoto University, he was challenged to clarify the mechanism of how R8 as a model of a cell penetrating peptide (CPP) entered cells.13) After fruitful discussions, I became interested in the mechanism of cellular entry of R8-conjugate materials not by endocytosis, but by a penetration like mechanism. After intensive study, we concluded that endocytosis is the key mechanism of R8-modified liposomes. However, the mechanism depended on the surface density of R8 on the liposomes.14) In the case of a high density of R8, micropinocytosis was the major entry mechanism but clathrin-mediated endocytosis was the mechanism for low density R8 liposomes. In the case of high density, liposomes seemed to escape from macropinosomes to reach nucleus, while, in the case of low density R8 liposomes, they seemed to be degraded after fusion with lysosomes with little escape from endosomes. This difference in intracellular trafficking seemed to me to be very interesting, since we can apply high density R8 liposomes for gene delivery. In parallel, we developed a new gene delivery system using R8 as a device to enhance cytosolic delivery as well as the cellular entry of pDNA, which was named as a MEND15) (Fig. 3). The transfection activity of the R8-MEND was much higher than we expected, since it could compete with the transfection efficiency of Adenoviral vectors.16) Since there was a clear difference in transfection efficiency between the R8-MEND and the K8-MEND, the advantage of R8 is not only a positive charge but also something related to arginine.17)

Our goal of the gene delivery study was to develop a DNA vaccine, since target cells are immune cells, which are easy to target by DDS, and a long duration of transfection activity was not required. We applied pDNA with the R8-MEND to dendritic cells (DC), which are considered to be a target for a DNA vaccine. Unfortunately, there was almost no transfection activity via R8-MEND, although the cellular uptake of R8-MEND in DC was sufficient.18) The nuclear membrane was the key barrier for pDNA with the R8-MEND and we tried many kinds of approaches to enhance the nuclear delivery of pDNA such as sugar-lipid conjugate and a short polyethylene glycol (PEG)-lipid conjugate.19,20)
A new strategy was developed to overcome nuclear membrane via membrane fusion. This strategy was referred to as a tetra-lamellar-MEND (T-MEND). We thought that the nuclear membrane could be overcome similar to the endosomal membrane, however, we needed two lipid membranes, since the nuclear membrane is a double bilayer membrane. To realize this strategy, we needed a new technology for packaging pDNA in a T-MEND and a new lipid composition that could induce membrane fusion with the nuclear membrane. A new packaging was developed by the Kogure group and lipid screening was done by the Akita group. Finally, these two technologies were established in our laboratory and combined to prepare a T-MEND that could overcome the nuclear membrane. The T-MEND succeeded in enhancing transfection activity by several hundred-fold compared to the original R8-MEND in a JAWS II cell line.21)
4.2. Introduction of the KALA Peptide for Nuclear Delivery of pDNAI believed that the T-MEND was an excellent idea as a non-viral gene delivery system, however, it could not induce antigen presentation in DC. We are disappointed with this result and our idea seemed to be exhausted. A few years later, a breakthrough was made by the introduction of the KALA peptide as well as cytosine-phosphate-guanine (CpG) free pDNA and a negative N/P ratio. These three conditions allowed transfection activity in DC to be induced sufficiently and major histocompatibility complex (MHC) class I antigen presentation was finally induced in DC.22) According to our hypothesis, the pDNA delivered to the nucleus should be increased compared to R8-MEND, however, there was no such difference between the R8-MEND and the KALA-MEND in the nuclear delivery of pDNA. It was surprising that a three order of magnitude difference came from after nuclear delivery processes such as transcriptional and translational process, which is similar to what was found in comparison between Adenovirus and Lipofectamin. This result seemed to be very interesting, however, the underlying mechanism was unclear. The KALA-MEND appeared to be a good candidate as a gene delivery system for DNA vaccines, however, it was difficult to convince Pharmaceutical Companies to move to clinical translation, since it does not work in vivo. They required something from us that works in vivo, otherwise no clinical translation.
4.3. BCG ProjectDr. Ken Sato is my old friend since senior high school. He is a medical doctor at Tsukuba University Hospital. I had an opportunity to be invited to present my research in their laboratory seminar. After the seminar, Professor Akaza was excited and proposed a research collaboration. He wanted to cure bladder cancer with BCG, not a live BCG but immunostimulative components of BCG using R8-MEND. According to his hypothesis, delivering BCG components into bladder cancer cells, which activate the immune system of patients and cure bladder cancer by itself is sufficient. We used BCG cell walls (BCG-CW), which consists of long chain fatty acids (mycolic acid) and different sugar molecules such as mycolyl arabinogalactan-peptidoglycan complex, lipoarabinomannan, lipomannan and phosphatidylinositol-(oligo) mannosides.23) We found a way to encapsulate BCG-CW into R8-liposomes with diameters less than 200 nm. R8-liposomes with BCG-CW were taken up by DC via endocytosis and stimulated DC by the expression of CD80, CD86 and MHC class II. Interleukin (IL)-12 was also induced. Antitumor activity was also obtained with a mouse bladder cancer cell line MBT by enhancing the tumor uptake of BCG CW encapsulated in R8-liposomes.24) The collaboration was going well and clinical applications were being considered. The BCG company proposed to use BCG-CWS rather than BCG-CW based on a purification view point. We then faced a serious problem in packaging BVG-CWS into R8-liposomes due to different physio-chemical properties. It was not easy to obtain small sized nanoparticles with a small PDI. However, we proceeded in re-establishing a method for packaging BCG-CWS into R8-liposomes and evaluated antitumor activity in a more realistic tumor model, a BBN-induced rat urinary bladder carcinoma model.25) As a result, rats receiving R8-liposome-BCG-CWS intravesically showed significantly inhibited numbers of tumors, especially those of simple hyperplasia, in comparison with the control rats.25)
We were interested in the hypothesis of anti-bladder tumor activity of BCG-CWS, which should be internalized by bladder tumor cells to the activate immune system. It was shown in MTB cells that had been inoculated in mice that internalization by MBT is essential for inducing an antitumor effect of BCG-CW encapsulated in R8-liposomes using the LEEL method.26,27) We were then challenged by the usefulness of the intravenous administration of BCG-CWS in R8-liposomes, since live BCG cannot be administered intravenously due to infection-induced problems. As we expected, when BCG-CWS was administered intravenously, antitumor activity was observed by inducing significant cytotoxic T lymphocytes,28) which could be attributed to uptake by DC in the spleen rather than B-cells.29)
4.4. MITO-PorterWhen we consider the importance of intracellular trafficking, endosomal escape is the most important step in targeting every organelle such as the nucleus, mitochondria, cytosol, etc. The nucleus is the most important organelle for gene delivery and the it is very difficult for pDAN to pass through the nuclear membrane, since the nuclear membrane is made up of two lipid bilayers and nuclear pore is too small for condensed pDNA to penetrate. In parallel, we started developing a system for delivery molecules to mitochondria, which, like the nucleus, also consists of two lipid bilayers. MTS, a mitochondria targeting signal, can be applied to enhance proteins to penetrate the mitochondrial membrane via a hole consisting of TOM (translocator of outer membrane) complex. Unfortunately, the utility of MTS is not wide enough to allow pDNA to transfer into mitochondria. The situation is similar with the nuclear delivery of pDNA. Therefore, we used membrane fusion as a key mechanism for overcoming the mitochondrial membrane as T-MEND did. Various lipid compositions were screened to enhance the fusion between nanoparticles and mitochondrial membrane using isolated mitochondria.30) PA or SM was identified as the optimalt lipid for fusion with the mitochondrial membrane in combination with dioleoylphosphatidylethanolamine (DOPE) and R8. These lipid compositions were referred to as MITO-Porters.30) Using deoxyribonuclease (DNase) as the cargo, we confirmed that the MITO-Porter can deliver a cargo into the matrix of mitochondria.31,32) It was not easy to convince mitochondrial researchers that the MITO-Porter can deliver a cargo into the matrix of mitochondria as in the case of the delivery of pDNA to the nucleus via membrane fusion. We selected antisense oligo (ASO) which is a good model compound as a marker for mitochondrial delivery, since ASO should be delivered to the matrix of mitochondria and exert gene silencing. After optimization of the MITO-Porter, ASO could be successfully delivered to mitochondria so that mRNA was decreased, proteins were also reduced and membrane potential was also decreased.33) In parallel with development of the MITO-Porter, we were challenged to make a reporter gene to mitochondria, since the lack of a reporter gene to mitochondria is one of the most critical reasons for the delayed development of mitochondrial delivery systems.34–36) Since the translational mechanism is different between mitochondria and the nucleus, we needed to design a DNA sequence that was compatible for the mitochondrial translational system.
Mitochondrial gene therapy was achieved via the MITO-Porter by encapsulating mitochondrial mRNA/ribosomal RNA (rRNA)/tRNA under the mutated mitochondrial gene. In the case of fibroblasts from a Leigh syndrome patient with a T10158C mutation in the mtDNA coding the ND3 protein, a component of the mitochondrial respiratory chain complex I, normal ND3 protein-encoding mRNA was delivered to mitochondria via the MITO-Porter as a therapeutic RNA.37) The mutation rate was reduced from 80 to 10% a dose-dependent manner and the maximal mitochondrial respiratory activity was increased from 65 to 90 OCR (pmol/min). In the case of primary cultured skin fibroblasts from a patient with a G625A mutation in mitochondrial tRNAPhe, wild-type mitochondrial pre-tRNAPhe was encapsulated into a MITO-Porter and transfected to diseased cells. As a result, the mutation rate of tRNAPhe reduced from 80 to 40%, relative mitochondrial respiratory capacity increased almost 50% compared to the control, the relative ATP production level increased more than 30% compared to the control.38) In the case of fibroblasts from patients with an A1555G point mutation in mitochondrial DNA coding 12S rRNA, wild-type rRNA (12S) as a therapeutic RNA was transfected using a MITO-Porter. The ratio of mutant rRNA to total rRNA decreased dose-dependently from 90 to 15%, while Lipofectamine RNA iMAX failed to decrease the mutation ratio to less than 50%. The maximal mitochondrial respiratory activity was increased by more than 80% compared to the control.39) These challenges will accelerate the clinical translation of mitochondrial gene therapy in the near future.
It was unexpected for me to create another laboratory for active targeting, which was made by the Gaisan Project (estimated budget proposed to the government from the university). I did not know this budget well until the Dean Yokozawa directed me to prepare a draft of an application for Gaisan Project. This project is not based on an individual researcher but on the overall University. After two failures of hearing by the government, a new Gaisan Project on “An innovative drug delivery system to target vasculature tissue selectively” was born and I created another Laboratory of Innovative Nanomedicine (Fig. 4). I was doing my best to compete with Professor Kataoka by increasing the numbers of graduate students, especially Ph.D. course students. However, it was not an easy task to create a second laboratory with highly motivated, excellent faculty members as well as graduate students. This project started in April 2009 and should have been over in 2013 (5 years), however, this project is still running after 15 years. Important research activities are introduced from this Gaisan Project.

Tumor tissue is one of the most heavily examined tissues in the field of DDS and EPR-effect (enhanced permeability and retention effect) has been the gold standard as a targeting strategy via passive targeting,40) however, a clear difference was observed between mice and humans in the effect of liposomal doxorubicin based on EPR-effect,41) which should be kept in mind when we move to clinical translation.42)
5.1.1. Dual Ligand SystemTo target tumor endothelial cells specifically and extensively, we developed a dual ligand system, which contains a specific ligand (NGR) to CD13 on the surface of angiogenic endothelial cells and a cell penetrating peptide (Rn). The number of arginine (n) units was optimized in in vitro condition at 4 and it was found that the cellular uptake of the R4-modified liposomes could be sufficiently increased by PEGylation. In addition, there was a synergistic effect in the enhanced cellular uptake between NGR and R4.43) The antitumor effect of encapsulated doxorubicin (DOX) was evaluated against renal cell carcinoma (RCC) cells which express CD13 that were and inoculated in BALB/cAJcl-nu/nu mice. A dual ligand system with DOX induced a significant as well as synergistic antitumor effect, while the single ligand (NGR or R4) failed to induce an antitumor effect.44) Another combination between cyclic RGD and R8 was also constructed to induce synergistic interactions.45)
5.1.2. cRGD-MENDThe effect of the size of the cRGD modified PEG-liposomes was examined for cellular binding as well as the antitumor effect of encapsulated DOX. A significant enhancement was observed in the binding affinity (11-fold) as well as the antitumor effect of the encapsulated DOX for large sized (300 nm) cRGD-PEG-liposomes compared to the small sized (100 nm) cRGD PEG-liposomes.46) RNA interference (RNAi)-mediated gene knockdown and anti-angiogenic therapy of RCC was evaluated using a cRGD-modified MEND that included YSK05 as an ionizable cationic lipid which was developed in our laboratory.47) vascular endothelial growth factor receptor (VEGFR)2 was chosen as a therapeutic gene and 3 mgsiRNA/kg was encapsulated in the cRGD/YSK-MEND and administered 3 times at a dose of 3.0 mgsiRNA/kg. Tumor volume was reduced to 30% of the control and tumor vessel area was also reduced significantly.47) In collaboration with Professor K. Hida, we chose biglican as a target gene to be expressed on the surface of tumor endothelial cells and to play important roles in tumor progression and metastasis.48) siRNA against biglycan was encapsulated into the cRGD/YSK-MEND and administered to mice with OS-RC2 cells. A 3 mgsiRNA/kg dose against biglycan with the cRGD/YSK-MEND was administered intravenously twice a week for 3 weeks and a 50% gene silencing was achieved as well as a 40% decrease in tumor volume.49)
5.1.3. Remodeling the Tumor MicroenvironmentIt was proposed that altering the tumor microenvironment would improve the intratumoral distribution of nanocarriers by decreasing extracellular matrix (ECM) molecules composed of type 1 collagen, glycosaminoglycans, proteoglycans, etc.50) We attempted to change the tumor microenvironment so that nanoparticles would be distributed homogenously in tumor tissues. The VEGF receptor 2 in tumor endothelial cells was targeted for silencing by siRNA via the cRGD/YSK-MEND in OS-RC2-bearing mice. After the inhibition of VEGFR2 in TEC, the intratumoral distribution of nanoparticles was more homogenous and the antitumor effect of DOX in liposomes was also enhanced significantly.51) Remodeling of the tumor microenvironment was evident by increased pericytes, increase in tumor blood flow rate, decreased hypoxia and decreased Type 1 collagen α1.51)
5.1.4. Active Targeting to Tumor Cells in VivoEpCAM, an epithelial cell adhesion molecule, was chosen as a target of tumor cells and a peptide ligand was developed via a RaPID system in collaboration with Professor Suga. The EpCAM-targeted MEND (ET-MEND) was developed to deliver siRNA to tumor cells via the high affinity of this ligand as well as prolonged blood circulating time. As a result, significant silencing of the PLK1 gene was achieved by the ET-MEND in Hep3B bearing mice, while little silencing was observed in the case of the PEG-MEND, although a longer circulation time was obtained for the PEG-MEND than that of ET-MEND.52) We were also challenged to cure hepatocellular carcinoma (HCC), where stroma-rich microenvironment hinders the in vivo delivery of nanoparticles. Ultra-small lipid nanoparticles (usLNPs) were designed to co-deliver a cytotoxic drug, sorafenib (SOR) and siRNA against Midkine gene (MK-siRNA) to HCC in mice. The optimized usLNPs with diameters of 60 nm with a HCC selective ligand (SP94 peptide) could induce gene silencing at a dose of 0.1 mg/kg and a significant antitumor effect of SOR was observed at a dose of 2.5 mg/kg, which was achieved by a synergistic interaction between SOR and MK gene silencing.53) The sensitivity of HCC against SOR was increased by silencing the MK gene.54)
5.2. LiverThe liver is a major organ for clearing nanoparticles from the blood circulation and is composed of hepatocytes, liver endothelial cells, Kupfer cells, hepatic stellate cells, etc.
5.2.1. HepatocytesIn the case of lipid nanoparticles (LNP) composed of ionizable cationic lipids (iCL), the surface the PEG-lipid was designed to be dissociated from the LNP within a few minutes after introduction into the blood circulation, the apolipoprotein E (ApoE) proteins are then bound on the surface of the LNP, which is recognized by low density lipoprotein-receptors (LDLR) on the surface of hepatocytes. In this case, the optimum pKa of LNPs in silencing genes was at 6.25 in most cases. We were interested in the initial disappearance phase of the LNPs and found that ApoE bound LNPs are first rapidly recognized by the heparin sulfate proteoglycan (HSPG) on the surface of hepatocytes and are then transferred slowly to LDLR.55)
5.2.2. Liver Endothelial CellsThe ApoB-100 segment RLTRKRGLK was introduced as a ligand for targeting liver endothelial cells (LEC). Peptide modified PEG-liposomes were found to selectively accumulate in LEC, which was inhibited by unlabeled peptide modified PEG-liposomes, while no inhibition by cationic liposomes occurred.56) LEC can also be targeted by an LNP composed of YSK05 and YSK12-C4 with pKa at 7.15 without ligands,57) which was artificially designed by a synthesized pKa between YSK05 (pKa = 6.50) and YSK 12-C4 (pKa = 8.00). Based on this method, we were able to synthesize the target pKa based on the combination of two kinds of pKa, one being above, the other being a lower pKa.
5.3. Spleen5.3.1. R8/YSK-MENDWe have been independently developing a gene delivery system using R8 peptides as CPPs and YSK lipids/CL4H6 as iCLs. Determining whether CPPs and iCL can interact synergistically was an interesting challenge. As we expected, there was significant synergism between a R8 peptide and a YSK05 lipid in gene expression in in vitro conditions.58) Synergism was induced when a low density of R8 peptide was introduced in the inner core with the pDNA with the YSK lipid in the outer layer as a two-step coating strategy. This system was applied to mice under in vivo conditions and it was found that the double-coated R8/YSK system could induce a remarkable transfection activity specifically in the spleen with little activity in the liver.59) The effect of a double-coating method increased the transfection activity by more than two order of magnitudes, which could induce antitumor activity against EG7 ovalbumin (OVA) tumor mice model.
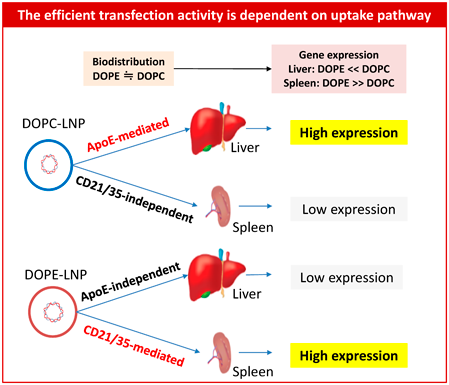
5.3.2. 1,2-Dioleoyl-3-dimethylammonium Propane (DODAP)-LNPWhen we use a DODAP as an iCL, the transfection activity of the DODAP/DOPE-LNP in spleen was increased by more than 1000-fold depending on the ratio of DODAP/DOPE being used. It was also found that the increased transfection activity in spleen was dependent on a complement receptor-mediated uptake pathway especially in B-cells.60) The synergistic increase in the transfection activity of the DODAP-LNP in spleen and liver was also investigated. It was found that the DODAP/DOPE-LNP induced increased transfection activity in the spleen by more than 10000-fold compared to the DODAP/DOPC-LNP that acted via a complement receptor-mediated uptake pathway. On the other hand, DODAP/DOPC-LNP induced increased transfection activity in the liver via ApoE-dependent pathway (Fig. 5). It was also found that these enhanced transfection activities result from translational processes.61)
We expected to extend the targetable tissue via the nanoparticulate system to organs other than liver, spleen and tumor tissue, since endothelial cells in each tissue are a high hurdle to overcome for nanoparticles to penetrate. Under such circumstances, we were challenged to selectively target adipose endothelial cells tissue by using a peptide ligand, KGGRAKD, which was recognized by prohibitin expressed on the surface of adipose endothelial cells.62) By introducing this peptide ligand at the edge of the PEG, prohibitine-targeted nanoparticles (PTNP) were designed in vitro63) and was extended to in vivo conditions. After optimization of the ligand condition on the surface of the nanoparticles, PTNP could successfully target adipose endothelial cells under in vivo conditions. By encapsulating cytochrome-C into PTNP, diet induced obesity was prevented by PTNP in a dose-dependent manner.64) A comparative study was also performed between PTNP and a conjugate of a proapoptotic peptide and a targeting peptide. Clear advantage was shown for PTNP against a conjugate system against diet induced obesity mice.65) It was surprising that the killing of endothelial cells can induce anti-obesity not only in adipose tissue but also in the liver, muscle, etc. These results suggest that a new mechanism underlies the targeting of adipose endothelial cells for anti-obesity.
5.5. Lung5.5.1. IRQBased on our finding that the peptide (IRQRRRR) serves as a targeting ligand for caveolae-mediated endocytosis, we examined the role of the IRQ peptide in the in vivo distribution in mice.66) It was found that IRQ-peptide could target lung endothelial cells 20-fold more efficiently than PEG-liposomes in mice. After optimization the IRQ-MEND to overcome the PEG-dilemma, transfection activity was improved more than 5 orders of magnitude.66)
5.5.2. GALAIt was serendipity for us to find that the GALA peptide had the ability to target lung endothelial cells, since we were using the GALA peptide as an endosomal escape device.67) The ability of the GALA peptide to target lung endothelial cells was much higher than we expected and lung accumulation was measured in vivo using real time confocal laser scanning microscopy in collaboration with Professor Kataoka.67) It was also found that the GALA-MEND can reach lung epithelial cells as well as lung endothelial cells via a caveolae mechanism.68) The GALA-MEND was then optimized in combination with iCL (YSK05), which could induce silencing efficiency up to 40-fold. This required the fine tuning of the helper lipid, cholesterol density, as well as GALA density.69) The GALA-system can be usde to deliver pDNA to lung endothelial cells and a 1000-fold increase in transfection activity was achieved by optimization.70)
5.5.3. Polyester LipomersA combinatorial library of polyester lipomers was designed based on the renewable monomer ε-decalactone (ε-DL), via organocatalytic ring-opening polymerization for delivering mRNA in collaboration with Professors Sato T and Isono T.71) After in vivo screening, we found that nanoparticles based on this library had a tendency to accumulate in the lung by escaping from the liver and spleen uptake.71)
It was very surprising to us that DLin-KC2-DMA enhanced gene silencing efficiency in the liver by two orders of magnitude compared to DLin-DMA72) in 2010, since there was little space in the case of siRNA delivery to the liver due to the limited factors available for determining overall efficiency. More than 80% of the dose is delivered to the liver in most cases, therefore there is only a 20% increase left. Endosome escape efficiency is considered to be the most important determinant of the efficiency of siRNA delivery and we have estimated 70% was achieved in the case of the R8/GALA-MEND of which the ED50 is 1 mg/kg, the same as that of DLinDMA. Therefore, we were thinking that only 44% (1–0.8*0.7) would be the maximum value for the efficiency to be increased, however, DLin-KC2-DMA increased 100-fold, which seemed impossible to achieve based on improvements of our strategy.
6.1. YSK05We followed the concept of iCLs and were surprised to see the efficiency of YSK05, which is a newly designed structure of iCL in our laboratory.73) The ED50 of siRNA to silence the FVII gene in the liver was enhanced by 17-fold (0.06 mg/kg) compared to that of conventional cationic lipid (1 mg/kg). In collaboration with Dr. Kohara who designed siRNAs to silence the viral gene of hepatitis C virus (HCV), we succeeded in silencing the viral gene in chimeric mice infected HCV.74) Two administrations of siRNA with the YSK05-MEND at a dose of 1 mg/kg succeeded in curing HCV infected chimeric mice.74) The blood level of HCV RNA was reduced for 2 weeks.
6.2. YSK13We also attempted to cure hepatitis B virus (HBV) which is more difficult to cure than HCV. It was difficult for YSK05 to cure HBV due to appearance of toxicity. The YSK13 which is an improved type of iCL with an ED50 at 0.01 mg/kg for FVII. HBV infected chimeric mice with humanized liver recovered after a single administration of the YSK13-MEND at a dose of 5 mg/kg, which reduced plasma HBV antigens for 2 weeks.75) Since YSK05 and YSK13 are novel structures, we submitted patent protection for their use from Hokkaido University, however, overlapping of lipid structures were found later with a Big Pharma patent and we had to abandon our patents.
6.3. Novel Lipid Library of iCLsWe recognized the importance of a chemical library of iCLs to identify the best efficiency. By changing not only the hydrophobic lipid tail structure but also the hydrophilic head structure, increased the combinations of new iCLs derivatives76) (Fig. 6) and CL4H6 was identified to have the best silencing efficiency with an ED50 at 0.0025 mg/kg in the liver, which is better than that of MC3, previously thought to be a gold standard iCL. Based on the newly constructed iCLs library, we were able to collect insights from a structure–activity relationship (Fig. 7).

Figure 1 of reference 76 was partly modified.

CL4H6 was applied to mRNA delivery by optimizing the lipid composition based on the design of experiments (DoE) and B-13 (CL4H6/SM/Chol/PEG = 60/5/35/1.5) was identified as the optimized condition for mRNA delivery. mRNA with B-13-LNP can induce hEPO proteins in the blood and can transfect mRNA more efficiently than MC3 for Nluc as well as mCherry expression in liver.77)
6.3.2. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Associated (Cas) DeliveryWe also attempted to deliver the CRISPR/Cas system with the CL4H6-LNP. A lot of experiments were required to determine the suitable microfluidics conditions to prepare CRISPR/Cas9 ribonucleoprotein (RNP) containing LNP, since RNP can be inactivated in acidic conditions as well as in hydrophobic conditions such as ethanol. After establishing the correct microfluidic conditions, RNP complexed with single-stranded oligonucleotide (ssODN) was loaded efficiently into the LNP, which was optimized to knock out the GFP gene using DoE and B-4 (CL4H6/DOPE/chol/PEG = 40/20/40/2) and B-9 (CL4H6/DOPE/PEG = 50/50/2) were identified as the optimized lipid conditions.78) These LNPs could induce the knock down of the enhanced green fluorescent protein (EGFP) gene by over 95% at 5 nM of RNP. The gene substitution from EGFP to BFP was generated in a concentration dependent manner and reached 23% at 5 nM of RNP. The Cas9 RNP-loaded B-9-LNPs inhibited HBV DNA and cccDNA by approximately 60 and 80%, respectively. AAV2 also inhibited both HBV DNA and cccDNA, however, the inhibitory effect was lower than that of LNPs.78) Sequence-specific RNP-ssODN complex formation was important for functional delivery of RNP and the melting temperature (Tm) between sgRNA and ssODN had a significant effect on in vivo genome editing. Tm optimized formulation achieved 70% transthyretin knockout at the DNA level as well as 80% at the protein level.79)
6.3.3. Activated Hepatic Stellate CellsA selective targeting system for activating hepatic stellate cells (aHSC) was developed without a targeting ligand based on a library of iCL. Based on in vivo screening, the mRNA was delivered selectively and efficiently. The optimized LNP was identified as CL15A6 as iCL, DOPE as the helper lipid, a diameter of 90 nmz, pKa at 7.25. The CL15A6-LNP could target aHSCs via PDGFβR-mediated endocytosis in fibrotic liver mice, while accumulated in the spleen in healthy mice.80) This system was applied to cure liver fibrosis by the co-administration of siRNAs against smothend homolot (SMO) and transforming growth factor beta 1 (TGFβ1) in CL15H6-LNPs. Five injections of a cocktail of siRNAs at a total dose of 0.5 mg/kg allowed the reprogramming of aHSC to qHSC (quiescent HSC) with subsequent reversal of liver fibrosis and restoration of the normal liver performance in mice.81)
The paper was long when I was writing, but it now seems short now that I am finished writing. The journey of how to create an artificial gene delivery system that functions as efficiently as a viral vector was a lengthy task and we needed to acquire elegant and expensive equipment to solve the problems that we faced regarding intracellular trafficking as well as biodistribution. Our journey was exciting and interesting, however, the end goal has not yet been reached. I would like to start this research again, if I had enough time as well as enough Grants and people to do it together. It seems to me that 25 years of my research in Hokkaido University was so short that it is like a dream. I have enjoyed my life by concentrating on research that I found to be interesting, exciting as rewarding. There are so many aspects which I have not been able to examine yet and I hope they will be investigated intensively by young researchers.
In addition to the scientific research, we have some products which have the potential for being translated into the clinic in collaboration with venture companies, chemical companies and pharmaceutical companies in Japan as well as overseas. My hopes are that they will contribute to innovative nanomedicines which will emerge in future.
I would like to express my thanks to Professor Yuichi Sugiyama who is my mentor and who nominated me for the Award of the Pharmaceutical Society of Japan. I also wish to thank all the colleagues who worked together in the Laboratory for Molecular Design of Pharmaceutics and the Laboratory of Innovative Nanomedicine, especially Professor Hiroyuki Kamiya, Professor Hidetaka Akita, Professor Kentaro Kogure, Professor Hiroto Hatakeyama, Professor Yuma Yamada, Associate Professor Takashi Nakamura, Assistant Professor Yusuke Sato, Specially Appointed Assistant Professor Mahmoud Younis, Lecturer Yu Sakurai, Professor Kazuaki Kajimoto, Professor Ikramy Khalil, Associate Professor Mamoru Hyodo, Dr. Yasuhiro Hayashi, Professor Kyoko Hida, Professor Manabu Tokeshi and Professor Kazunori Kataoka. I would also like to express my sincere appreciation to all the students who studied together in our laboratories. Finally, I would like to thank Dr. Milton Feather for his English editing.
Hideyoshi Harashima has a patent JP7202009 licensed to JSR Corporation and a patent EU18816591.4 licensed to Merck KGaA.
This review of the author’s work was written by the author upon receiving the 2023 Pharmaceutical Society of Japan Award.