Abstract
Huntington’s disease (HD) is a hereditary neurodegenerative disease that involves an expansion of the CAG repeats of the Huntingtin (HTT) gene, but the disease onset and progression do not necessarily correspond to the extent of CAG repeats. Decreased mitochondrial complex II activity has also been reported to be closely associated with disease pathogenesis. Here, we examined the mechanism of cell death induced by 3-nitropropionic acid (3-NP), a mitochondrial complex II inhibitor, using striatal cells (STHdhQ111 cells) derived from HD model mice with mutant HTT carrying the CAG repeat extended. Treatment with 3-NP (5 mM) enhanced cell death in STHdhQ111 compared to STHdhQ7 cells with normal HTT. Ferrostatin-1, an inhibitor of ferroptosis, and deferoxamine, an iron chelator, markedly inhibited 3-NP-induced cell death in both the STHdh cell lines. On the other hands, cell death was not abrogated by a broad-spectrum caspase inhibitor, Z-VAD-FMK, indicating that this cell death was caspase-independent. Cell death caused by 3-NP is suggested to be due to ferroptosis. Furthermore, 3-NP-induced cell death was markedly inhibited by GSK2795039, a reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) inhibitor, suggesting that cell death is mainly mediated by intracellular superoxide anion (O2−) production through NOX2. Furthermore, a mitochondria-targeted superoxide dismutase mimetic (Mito-TEMPO), partially inhibited 3-NP-induced cell death, suggesting that O2− production in the mitochondria is partially responsible for cell death. These results indicate that 3-NP-induced cell death in the STHdhQ111 cells is caspase-independent, non-apoptotic, and that ferroptotic cell death is mainly induced via NOX2 activation.
INTRODUCTION
Huntington’s disease (HD) is a hereditary neurodegenerative disease, where the Huntingtin (HTT) gene with an expanded CAG repeat sequence has been identified as the causative gene.1) The mutant HTT protein (mHTT), with abnormal aggregation, interacts with various intracellular molecules and can induce dysfunction and cell death in the striatum and cortical neurons. However, the detailed mechanisms of HD onset and progression have not yet been fully elucidated. In particular, even among patients with the same CAG repeat numbers, there is variability in the onset of the disease, degree of symptoms, and progression rate, implicating that genetic background and environmental factors other than mHTT are profoundly associated in the onset and progression of the disease.2) The major symptoms of HD are involuntary movements, psychiatric disorders, and impairment of cognitive function, which appear primarily in adults but can be seen in across age groups.3) Despite clinical trials being conducted to develop direct therapeutic interventions against mHTT, no effective drugs have been developed yet.4) The transgenic HD mouse model with CAG repeats (R6/2 mice),5) chemical-induced HD model,6) and administration of 3-nitropropionic acid (3-NP), an inhibitor of mitochondrial complex II, have been widely used to understand the pathogenesis of HD. It has been reported that the R6/2 HD model mice are susceptible to 3-NP-induced metabolic dysfunction and striatal damage.7) In our previous study, we reported that 3-NP treatment enhanced cell death in STHdhQ111 striatal cells (containing homozygous mutant HTT with CAG repeat expansion) in comparison with STHdhQ7 striatal cells, suggesting that intracellular reactive oxygen species (ROS) is associated with the induction of cell death.8) Recently, it has been suggested that ferroptosis is involved in HD-related cell death.9) Ferroptosis is an iron-dependent cell death identified recently, which includes increased lipid peroxidation, iron ion accumulation, and alterations of iron-related proteins. Ferroptosis has also been reported in patients with HD and R6/2 HD model mice.10–12) Furthermore, deferoxamine, an iron chelator, has been reported to improve motor dysfunction and neurological abnormalities in R6/2HD mice.12) Ferroptosis involves excess lipid peroxidation, but little is known about its signaling and effector molecules.
In this study, we investigated the involvement of intracellular ROS (NADPH oxidase) in ferroptosis following treatment of STHdhQ111/STHdhQ7 cells with 3-NP.
MATERIALS AND METHODS
Cell CultureThe mouse STHdhQ7 (wild-type HTT expression) and STHdhQ111 (mutant HTT expression) striatal derived cell lines were purchased from the Coriell Institute for Medical Research (Camden, NJ, U.S.A.).13) These cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal calf serum (FCS) in a humidified atmosphere at 33 °C with 5% CO2 as described previously.8)
Cell Viability and Cell Death AssayCell viability and death were evaluated according to our previous study.8) Briefly, STHdhQ7 and Q111 cells were seeded at a density of 3 × 103 cells/well in 96-well plates (BD Biosciences, Franklin Lakes, NJ, U.S.A.) and incubated under 5% CO2 and 33 °C for 24 h. Deferoxamine mesylate (an iron chelator; Sigma-Aldrich, St. Louis, MO, U.S.A.), Ferrostatin-1 (a potent and selective inhibitor of ferroptosis; Sigma-Aldrich), GSK2795039 (an inhibitor of NOX2; Sigma-Aldrich), a mitochondria-targeted superoxide dismutase mimetic (Mito-TEMPO; Cayman Chemical Company, Ann Arbor, MI, U.S.A.), NG-Nitro-L-arginine methyl ester hydrochloride [L-NAME: a non-selective inhibitor for nitric oxide synthetases (NOS); Wako Pure Chemical Corporation, Osaka, Japan], and Z-VAD-FMK (an irreversible pan-caspase inhibitor; Toronto Research Chemicals, Toronto, ON, Canada) were dissolved in dimethyl sulfoxide (DMSO; Nacalai Tesque), then diluted in phosphate-buffered saline (PBS) including 1% DMSO, and applied to the wells to be in the optimal concentration range with 0.1% DMSO. 2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, sodium salt (Carboxy-PTIO: a nitric oxide scavenger; Dojin Kagaku, Kumamoto, Japan) was dissolved in PBS and added to the wells ad per the optimal concentration range. 3-Nitropropionic acid (3-NP; Cayman Chemical Company) was also dissolved in PBS to a final concentration at 5 mM. After the medium was replaced with DMEM with 3% FCS, the aforementioned reagents were applied and cultured for 24 h. After that, cell viability was assessed using the Cell Counting Kit-8 (CCK-8; Dojin Kagaku) in accordance with the protocol of manufacturer. All measurements were conducted at 0 and 2 h after addition of the CCK-8 solution by using a multiplate reader (Thermo Fisher Scientific, Waltham, MA, U.S.A.) at 450/650 nm. Cell death was determined by Hoechst 33342 (Thermo Fisher Scientific) and propidium iodide (PI; Thermo Fisher Scientific) staining. Hoechst 33342 and PI were both added to the culture medium at the same time to final concentrations of 8.1 and 1.5 µM, respectively. The images were acquired with a Lionheart FX automated microscope (BioTek, Winooski, VT, U.S.A.) after incubation for 15 min. The number of PI-positive dead cells and the total cell number of Hoechst 33342-positive cells was automatically counted using the Gen5 software (BioTek), and the percentage of dead cells was calculated.
Remarks: The CCK-8 assay used for cell viability measures reduced nicotinamide adenine dinucleotide (NADH) levels, an intracellular dehydrogenase cofactor. NADH levels in cells reflect the energy metabolic activity of the cells. On the other hand, the percentage of dead cells was evaluated by fluorescent nuclear staining with PI (dead cells) and Heachst33342 (whole cells). PI is not permeable to the plasma membrane of living cells, but only penetrates the plasma membrane in dead cells (late apoptosis and necrosis) and intercalates into the nuclear DNA, emitting red fluorescence. Thus, the assessment of cell viability reflects a decrease in energy metabolism activity (decrease in NADH content), including in living cells that are not stained with PI. Since some cells that are disrupted beyond the threshold of cellular homeostasis, including their reduced energy metabolism, lead to cell death, no difference can be detected in the assessment of cell viability when the rate of dead cells is low, or when the inhibition rate against cell death is insufficient.
ROS AssayProduction of intracellular ROS was assessed using CM-H2DCFDA reagent (Thermo Fisher Scientific) according to our previous study.8) Treated cells were incubated with CM-H2DCFDA at 33 °C for 60 min. Fluorescence signals were measured at 495/527 nm using a Varioskan Flash 2.4 microplate reader (Thermo Fisher Scientific), and corrected with the Hoechst 33342-positive cell number.
Western BlottingCells were washed with PBS and lysed with a lysis solution (RIPA buffer; Sigma-Aldrich) containing protease inhibitors (P8340; Sigma-Aldrich) and phosphatase inhibitors (P5726, P0044; Sigma-Aldrich). The cell lysates were centrifuged at 12000 × g for 20 min and the protein concentrations in their supernatants were measured using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific). Lysates were dissolved in sodium dodecyl sulfate (SDS) sample loading buffer containing 2-Mercaptoethanol, separated on 5–20% gradient SDS-polyacrylamide gel electrophoresis (PAGE) gels (SuperSep (TM) Ace; FUJIFILM Corporation, Tokyo, Japan), and then transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; EMD Millipore Corporation, Billerica, MA, U.S.A.). Transfers were incubated in a blocking solution (Blocking One-P; Nacalai Tesque) for 30 min at room temperature and further incubated overnight at 4 °C with the primary antibodies (mouse anti-β-actin (1 : 2000; Sigma-Aldrich, catalog A2228), rabbit anti-ferritin antibody (1 : 1000; Abcam, Cambridge, U.K., catalog ab75973)). The membranes were then incubated with secondary antibodies (peroxidase-conjugated goat anti-mouse antibody (Thermo Fisher Scientific) or peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (Thermo Fisher Scientific)). Immunoblots were developed with chemiluminescence (ImmnoStar LD; Wako Pure Chemical Corporation), visualized and quantified using a chemiluminescence imager (Amersham Imager 680; Cytiva, Marlborough, MA, U.S.A.).
Statistical AnalysesAll data are expressed as mean ± standard error of the mean (S.E.M.). Statistical analyses were conducted with a statistical analysis software (IBM SPSS Statistics; IBM, Armonk, NY, U.S.A.). For statistical comparisons, Tukey’s test was used. p < 0.05 was considered statistically significant.
RESULTS
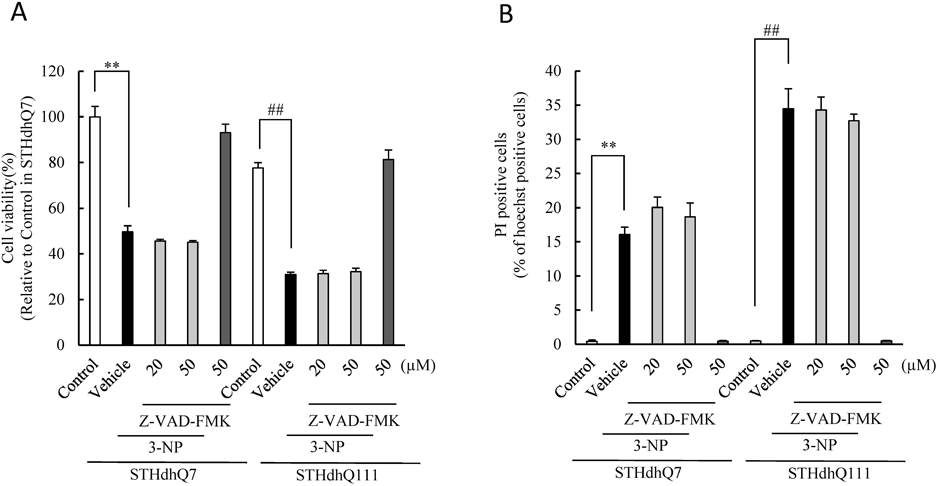
3-NP-Induced Cell Damage Was Not Inhibited by a Pan-Caspase Inhibitor in Striatal Cells with or without Mutant HTTIn a previous study, we reported that mouse striatum-derived STHdhQ111 cells expressing mutant HTT were more susceptible to 3-NP than STHdhQ7 cells with normal HTT, supported by the finding that caspase-3/7 activity was increased in STHdhQ111 cells but not in STHdhQ7 cells after 3-NP treatment.8) Interestingly, the increased caspase-3/7 activity was significantly inhibited by antioxidants.8) To clarify whether the cell death occurred through a caspase-dependent pathway, we tested the effects of a pan-caspase inhibitor (Z-VAD-FMK) (Fig. 1). As in our previous study,8) treatment with 3-NP (5 mM) reduced cell viability and increased cell death in both the STHdh cell lines and augmented them in STHdhQ111 cells compared to STHdhQ7 cells (Fig. 1). However, there was no evident effect of Z-VAD-FMK (20 and 50 µM) on the 3-NP-induced decrease in cell viability and increase in cell death in both the STHdh cell lines (Figs. 1A, B), suggesting that 3-NP-induced STHdh cell death may be mediated through a caspase-independent pathway.
Increased Ferritin Level in Mutant HTT Striatal Cells and 3-NP Treated Striatal Cells STHdhQ7Next, we examined whether ROS-induced STHdh cell death via the caspase-independent pathway is ferroptotic cell death. First, we investigated the expression of ferritin, a marker of iron accumulation by Western blotting (Fig. 2). In STHdhQ111 cells, the protein levels of ferritin heavy and light chains tented to be higher as compared to STHdhQ7 cells (Figs. 2A–C). Furthermore, 3-NP treatment significantly increased ferritin heavy chain levels in both the STHdh cell lines (Fig. 2B).
Ferrostatin-1 and Deferoxamine Treatment Reduces Cell Damage Post-3-NP Treatment in Striatal Cells with or without Mutant HTTAn inhibitor of ferroptosis, Ferrostatin-1, markedly suppressed the 3-NP-induced reduced cell viability and elevated cell death in both the STHdh cell lines (Figs. 3A, B). Furthermore, deferoxamine, an iron chelator, concentration-dependently inhibited the 3-NP-induced reduced cell viability and elevated cell death (Figs. 3C, D). These results suggest that 3-NP-induced cell death involves ferroptosis, mediated by free iron-mediated lipid peroxidation.
NOX2 Inhibitor Has a Protective Effect on 3-NP-Induced Cell Damage in Both STHdhQ111 and STHdhQ7 CellsIn our previous study, we showed that intracellular ROS production rather than mitochondrial ROS was the primary contributor to 3-NP-induced STHdh cell death.8) We hypothesized that intracellular ROS mediator NOX might be involved in cell death. We then investigated the role of NOX-mediated ROS production in cell death (Fig. 4) using a NOX2 inhibitor, GSK2795039. The treatment with GSK2795039 markedly inhibited 3-NP-induced cell death in both the STHdh cell lines (Figs. 4B, D), while the reduction in cell viability was significantly suppressed only in STHdhQ111 cells (Fig. 4C). These results suggested that intracellular superoxide anion (O2−) production via NOX2 is involved in 3-NP-induced cell death.
NOX2 Inhibitor Attenuates Intracellular ROS Production Induced by 3-NP in Both STHdhQ111 and STHdhQ7 CellsThe effect of NOX2 inhibitor on intracellular ROS production induced by 3-NP was determined (Fig. 4E). 3-NP treatment for 24 h significantly increased intracellular ROS production (Fig. 4E) in both cells. Treatment with GSK 2795039 inhibited their increases in ROS production in a concentration-dependent manner.
NOX2 Inhibitor Is Unaffected in Increased Ferritin Levels Induced by 3-NP in Both STHdh Cells with or without Mutant HTTThe effect of NOX2 inhibitor on the expression of ferritin, a marker of iron accumulation, were examined (Fig. 5). 3-NP significantly increased the expression of ferritin heavy chain and light chain in both cells (Figs. 5A–C). On the other hand, GSK 2795039 (5 µM) had no effect on the increased ferritin heavy chain and light chain in either cell (Figs. 5B, C). These results suggest that ROS production via NOX2 activation by 3-NP is not directly involved in iron accumulation.
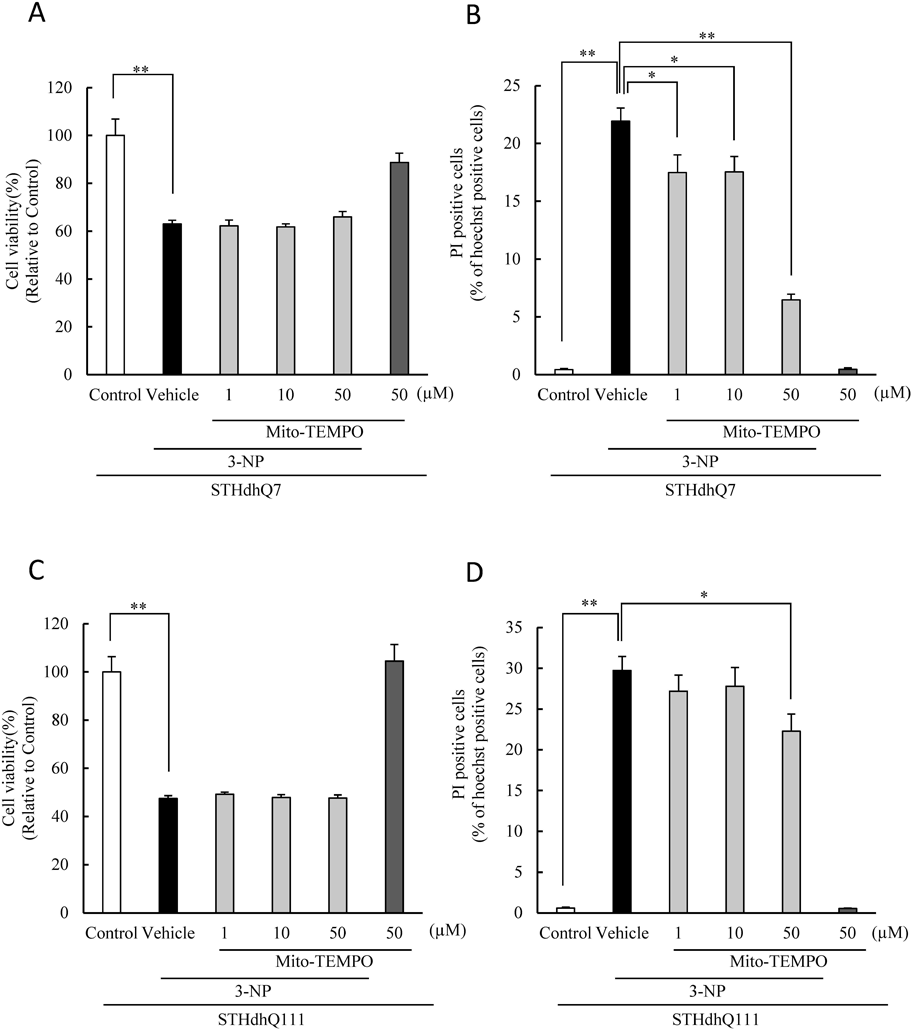
3-NP-Induced Cell Damage Is Ameliorated by a Mitochondria-Targeted Superoxide Dismutase, Mito-TEMPOCrosstalk between NOX activation and mitochondrial ROS production has been well documented.14) To investigate the involvement of mitochondrial ROS in 3-NP cell death, we examined the effect of Mito-TEMPO, a mitochondria-targeted superoxide dismutase mimetic (Fig. 6). Mito-TEMPO treatment partially inhibited 3-NP-induced cell death in both the STHdh cell lines only at a higher concentration (50 µM) (Figs. 6B, D). However, there was no evident effect on decreased cell viability (Figs. 6A, C).
A NOS Inhibitor, L-NAME, Failed to Inhibit 3-NP-Induced Cell Damage in Striatal CellsSystemic 3-NP administration in rats has been shown to exhibit HD-like symptoms, and NOS-mediated NO production has been suggested to be involved in HD.15,16) Therefore, we examined the action of L-NAME, an NOS inhibitor (Fig. 7), and found no evident effect of L-NAME (0.1, 1 mM) on the 3-NP-induced reduced cell viability and increaseed cell death of both the STHdh cells (Figs. 7A, B), suggesting that endogenous nitric oxide synthesis by NOS is not responsible for 3-NP-induced STHdh cell death.
A Nitric Oxide Scavenger, Carboxy-PTIO Ameliorates 3-NP-Induced Cell Damage in StriatalIt has been reported that 3-NPA generates NO directly as a donor to induce neuronal toxicity.15) Therefore, we investigated the effect of Carboxy-PTIO, an NO scavenger, on 3-NP-induced cytotoxicity (Fig. 8). Carboxy-PTIO inhibited 3-NP-induced cell death in both the STHdh cells in a concentration-dependent manner (Figs. 8B, D), while the decreased cell viability was significantly reduced in STHdhQ111 cells (Fig. 8C). These results suggest that 3-NP contributes in part as a direct NO donor.
DISCUSSION
HD is a neurodegenerative disease that is progressive, caused by mutations in the autosomal inherited HTT gene, leading to the CAG repeat expansion.1) Currently, there is no effective therapy to treat the progression of HD, while symptomatic treatment of motor dysfunction and psychiatric symptoms is the only available treatment. Therefore, to understand HD pathology, it is necessary to clarify the mechanisms underlying the pathogenesis and progression of the disease by using both clinical findings and animal models of HD pathology. The onset of HD pathology is a negative correlation with the CAG repeat number of mutant HTT, but its onset and degree of progression do not necessarily correspond to the number of CAG repeats, wherein other genetic backgrounds and environmental factors may be deeply involved.2,3) Notably, mitochondrial complex II–III activities were decreased in the striatum of HD patients,17) and systemic treatment with 3-NP, a mitochondrial complex II inhibitor was documented to produce HD-like phenotypes in the striatum of mice.6) Furthermore, it has been reported that the R6/2 HD model mice are susceptible to 3-NP-induced metabolic dysfunction and to striatal damage.7) In our previous study,8) we reported that STHdhQ111 cells with mutant HTT were more susceptible to cell death by treatment with 3-NP than wild-type STHdhQ7 cells and that intracellular ROS production was responsible for cell death. On the contrary, no obvious differences in cell survival activity and cell death were observed in STHdhQ111 cells as compared to STHdhQ7 cells under normal culture conditions, but an increase in the BAX/Bcl2 ratio (an index of pro-apoptosis) and a significant reduction in the p-Akt/t-Akt ratio (an index of cell survival activity) were observed. Furthermore, treatment with 3-NP enhanced caspase-3/7 activation in STHdhQ111 cells, but did not in STHdhQ7 cells, and antioxidants significantly inhibited this increase. These results suggest that 3-NP-induced cell death operates via the caspase-independent pathway in both STHdh cell lines, although activation of the caspase-3/7 pathway is susceptible to initiation only in STHdhQ111 cells.
Increased ROS production, lipid peroxidation, and iron ions have been reported in patients with HD.10,11) Similar changes in ROS production, lipid peroxidation, and iron-related proteins have been reported in R6/2 HD mouse models.11,12) Furthermore, deferoxamine, an iron chelator, improves motor dysfunction and neurological deficits in R6/2 HD mice.12) These findings strongly suggest the involvement of ferroptosis in cell death in HD pathogenesis. In the present study, ferrostatin-1, a ferroptosis inhibitor, and deferoxamine, an iron chelator, markedly inhibited 3-NP-induced cell death in both the STHdh cell lines, strongly suggesting that 3-NP-induced cell death is associated with ferroptosis. Furthermore, under normal culture condition (control) ferritin expression tended to be increased in STHdhQ111 compared to STHdhQ7. In Huntington’s disease model (R6/2) mice, iron accumulation in striatum, decreased iron regulatory protein (IRP-1/2) and increased ferroportin, which has iron efflux function, have been reported.12) In case of low intracellular iron levels, IRP binds to an iron responsive element (IRE) in the 5′ UTR of the untranslated region of mRNA for iron-related factors such as ferritin light chain, ferritin heavy chain, and ferroportin, suppressing their translation. On the other hand, ferritin and ferroportin are increased by inactivation and degradation of IRP by the increased intracellular iron. Thus, ferritin is thought to be increased in STHdhQ111. It has also been reported that ferroptosis inducers such as erastin and sorafenib activate the p62-Keap1-NRF2 pathway, which regulates the expression of antioxidant-related factors and ferritin heavy chain.18) In the present study, 3-NP treatment increased ferritin heavy chain, and the increase was marked at 24 h in both cell lines In a previous study using STHdhQ111, it was also reported that STHdhQ111 showed reduced antioxidant responsive element (ARE) activity regulated by NRF2.19) These findings suggest that STHdhQ111 is vulnerable to oxidative stress such as 3-NP treatment, because of constitutive iron accumulation in STHdhQ111.
ROS are upstream initiators of ferroptosis; however, possible sources of ROS include mitochondrial ROS and membrane-associated ROS derived from the NOX protein family. Therefore, we investigated the involvement of NOX in 3-NP cell death and found that GSK2795039, a NOX2 inhibitor, markedly suppressed 3-NP-induced STHdh cell death. Furthermore, intracellular ROS production was increased by 3-NP treatment in both cells, and GSK2795039 inhibited these increases in a concentration-dependent manner. On the other hand, the increase in 3-NP-induced ferritin production was not obviously inhibited by GSK2795039. These results suggest that ROS production by NOX2 activation is not responsible for iron accumulation itself, which induces ferritin, and is contributes additively and/or synergistically to lipid peroxidation, which is involved in downstream ferroptotic cell death. In contrast, Mito-TEMPO, an inhibitor of mitochondrial ROS, partially inhibited cell death at a high concentration (50 µM). These results suggest that NOX2-mediated production of O2− is the primary mediator of 3-NP-induced cell death, and that mitochondria-derived O2− is partially involved. However, further studies are needed to elucidate these precise mechanisms.
Systemic administration of 3-NP induces HD-like symptoms in rats, and NOS-mediated NO production is involved in its pathogenesis.15,16) In the present study, L-NAME, an NOS inhibitor, showed no evident effect on 3-NP-induced cell death, suggesting no involvement of endogenous NO in STHdh cells. In contrast, carboxy-PTIO, an NO scavenger, inhibited 3-NP-induced cell death in both the STHdh cell lines in a concentration-dependent manner. It has been reported that 3-NP directly generates NO as a donor for neuronal toxicity.15) These results suggest that 3-NP contributes in part as a direct NO donor, and that its contribution to cell death is implicated in peroxynitrite (ONOO−), a reaction product with O2−, or its nitrosylation to thiol bases.
CONCLUSION
We demonstrated for the first time that 3-NP-induced STHdh cell death is caspase-independent, and that it induces ferroptosis via NOX2-mediated O2− production, NO, and iron ion production. Furthermore, this cell death was enhanced in STHdhQ111 cells with mutant HTT.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) MacDonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72, 971–983 (1993).
- 2) Wexler NS, Lorimer J, Porter J, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. U.S.A., 101, 3498–3503 (2004).
- 3) Gusella JF, MacDonald ME. Huntington’s disease: the case for genetic modifiers. Genome Med., 1, 80 (2009).
- 4) Kwon D. Failure of genetic therapies for Huntington’s devastates community. Nature, 593, 180 (2021).
- 5) Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell, 87, 493–506 (1996).
- 6) Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem., 95, 1521–1540 (2005).
- 7) Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington’s disease. J. Neurochem., 71, 2642–2644 (1998).
- 8) Okada N, Yako T, Nakamura S, Shimazawa M, Hara H. Reduced mitochondrial complex II activity enhances cell death via intracellular reactive oxygen species in STHdhQ111 striatal neurons with mutant huntingtin. J. Pharmacol. Sci., 147, 367–375 (2021).
- 9) Mi Y, Gao X, Xu H, Cui Y, Zhang Y, Gou X. The emerging roles of ferroptosis in Huntington’s disease. Neuromolecular Med., 21, 110–119 (2019).
- 10) Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects. J. Neurol., 254, 1676–1683 (2007).
- 11) Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW, Ryu H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol., 121, 487–498 (2011).
- 12) Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH. Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PLOS ONE, 8, e77023 (2013).
- 13) Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum. Mol. Genet., 9, 2799–2809 (2000).
- 14) Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med., 51, 1289–1301 (2011).
- 15) Deshpande SB, Hida H, Takei-Io N, Masuda T, Baba H, Nishino H. Involvement of nitric oxide in 3-nitropropionic acid-induced striatal toxicity in rats. Brain Res., 1108, 205–215 (2006).
- 16) Kumar P, Kumar A. Protective effect of hesperidin and naringin against 3-nitropropionic acid induced Huntington’s like symptoms in rats: possible role of nitric oxide. Behav. Brain Res., 206, 38–46 (2010).
- 17) Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol., 39, 385–389 (1996).
- 18) Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology, 63, 173–184 (2016).
- 19) Jin YN, Yu YV, Gundemir S, Jo C, Cui M, Tieu K, Johnson GV. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLOS ONE, 8, e57932 (2013).