Abstract
We examined whether the α1L-adrenoceptor (AR), which shows low affinity (pA2 < 9) for prazosin (an α1-AR antagonist) and high affinity (pA2 ≈ 10) for tamsulosin/silodosin (α1A-AR antagonists), is involved in phenylephrine-induced contractions in the guinea pig (GP) thoracic aorta (TA). Intracellular signaling induced by α1L-AR activation was also examined by focusing on Ca2+ influx pathways. Tension changes of endothelium-denuded TAs were isometrically recorded and mRNA encoding α-ARs/Ca2+ channels and their related molecules were measured using RT-quantitative PCR. Phenylephrine-induced contractions were competitively inhibited by prazosin/tamsulosin, and their pA2 value were calculated to be 8.53/9.74, respectively. These contractions were also inhibited by silodosin concentration-dependently. However, the inhibition was not competitive fashion with the apparent pA2 value being 9.48. In contrast, phenylephrine-induced contractions were not substantially suppressed by L-765314 (an α1B-AR antagonist), BMY 7378 (an α1D-AR antagonist), yohimbine, and idazoxan (α2-AR antagonists). Phenylephrine-induced contractions were markedly inhibited by YM-254890 (a Gq protein inhibitor) or removal of extracellular Ca2+, and partially inhibited by verapamil (a voltage-dependent Ca2+ channel (VDCC) inhibitor). The residual contractions in the presence of verapamil were slightly inhibited by LOE 908 (a receptor-operated Ca2+ channel (ROCC) inhibitor) and strongly inhibited by SKF-96365 (a store-operated Ca2+ channel (SOCC) and ROCC inhibitor). Among the mRNA encoding α-ARs/SOCC-related molecules, α1A-AR (Adra1a)/Orai3, Orai1, and Stim2 were abundant in this tissue. In conclusion, phenylephrine-induced contractions in the GP TA can be triggered by stimulation of Gq protein-coupled α1L-AR, followed by activation of SOCCs and VDCCs.
INTRODUCTION
α1-Adrenoceptors (ARs), representative G protein-coupled receptors,1) are classified into three genetically distinct subtypes (α1A, α1B, and α1D).2) Stimulation of these three α1-AR subtypes with AR agonists has been reported to produce contractions in various smooth muscle tissues3–42) (Table 1). Cell lines overexpressing each α1-AR subtype show high affinity for the α1-AR antagonist prazosin (pKB > 9).43–46) However, in native smooth muscle (SM) tissues, the presence of an α1-AR showing low affinity for prazosin (pA2 < 9) was suggested, and this α1-AR type was proposed to be classified as α1L-AR, as opposed to the α1H-AR that shows high affinity for prazosin (pA2 > 9).43–46) Although many approaches have been used to identify the α1L-AR-encoding gene, it has not yet been found. Currently, α1L-AR is regarded as an α1A-AR-derived phenotype that is modified specifically in SM tissues. The evidence supporting this hypothesis is as follows: 1) α1L-AR shows high affinity for α1A-AR antagonists such as tamsulosin and silodosin (pA2 ≈ 10); 2) while α1L-AR characteristic binding sites can be detected using native SMs, the binding site characteristics change to the α1A-AR type using membrane fractions prepared by homogenization; and 3) in α1A-AR knockout mice, both α1L- and α1A-AR characteristics are not detected, while α1L-AR characteristics remain in α1B- and α1D-AR knockout mice.43–46) α1L-AR is abundant in the lower urinary tract and genital SMs and is substantially present in some vascular SM tissues19,28,32,38,39,47–65) (Table 1).
Table 1. α-Adrenoceptor (AR) Subtypes Involved in Various Smooth Muscle Contractions
| α1-AR Subtypes | Species | Smooth muscle tissues | References |
|---|
| α1A-AR | Human | Vas deferens | 3,4) |
| Prostate | 5) |
| Corpus cavernosum | 6) |
| Marmoset | Urethra | 7) |
| Dog | Superior tarsal muscle | 8) |
| Mesenteric artery | 9) |
| Pig | Urethra | 10) |
| Coronary artery | 11) |
| Rabbit | Thoracic aorta | 12) |
| Common iliac artery | 12) |
| Bronchus | 13) |
| Rat | Vas deferens | 14–17) |
| Prostate | 18) |
| Urinary bladder | 19) |
| Urethra | 20) |
| Cauda epididymis | 21) |
| Caudal artery | 22) |
| Renal artery | 22) |
| Renal interlobar arteries | 23) |
| Hamster | Ureter | 24) |
| Mouse | Urethra | 7) |
| α1B-AR | Human | Iliac artery branches | 25) |
| Internal mammary artery | 26) |
| Rabbit | Corpus cavernosum | 27) |
| Thoracic aorta | 12) |
| Common iliac artery | 12) |
| Cutaneous resistance arteries | 28) |
| Guinea pig | Spleen | 29) |
| Rat | Spleen | 14,15) |
| Tail artery | 30,31) |
| Mesenteric resistance artery | 22) |
| Portal vein | 32) |
| Mouse | Spleen | 33) |
| α1D-AR | Rabbit | Renal artery | 34) |
| Iliac artery | 34) |
| Rat | Vas deferens | 17) |
| Thoracic/Abdominal aorta | 22,31,35–38) |
| Superior mesenteric artery | 22,39) |
| Mesenteric artery | 31,37) |
| Pulmonary artery | 37) |
| Iliac artery | 22,40) |
| Carotid artery | 31) |
| Femoral artery | 22) |
| Mouse | Vas deferens | 41) |
| Thoracic aorta | 42) |
| α1L-AR | Human | Bladder neck | 47) |
| Prostate | 47) |
| Urethra | 47) |
| Vas deferens | 48) |
| Erectile tissue | 49) |
| Iris dilator muscle | 50) |
| Dog | Prostate | 51) |
| Urethra | 52) |
| Subcutaneous resistance arteries | 53) |
| Pig | Urethra | 54) |
| Rabbit | Bladder trigone | 55,56) |
| Urethra | 55,56) |
| Prostate | 56,57) |
| Mesenteric artery | 56) |
| Ear artery | 58) |
| Cutaneous resistance arteries | 28) |
| Iris dilator muscle | 50) |
| Guinea pig | Prostate | 59) |
| Aorta | 60) |
| Nasal mucosa vasculature | 61) |
| Rat | Urinary bladder | 19) |
| Superior mesenteric artery | 39) |
| Small mesenteric artery | 62) |
| Tail artery | 38) |
| Femoral artery | 63) |
| Portal vein | 32) |
| Mouse | Vas deferens | 64) |
| Prostate | 64,65) |
Regarding SM contractions mediated through α1H-ARs (α1A, α1B, and α1D), extracellular Ca2+ influx through Ca2+ channels plays a primary role similar to the contractions mediated through other drug receptors, and this is evidenced by the following findings: 1) α1A-AR-mediated, noradrenaline (NA)-induced contractions in rat vas deferens were elicited by Ca2+ influxes through voltage-dependent Ca2+ channels (VDCCs) as well as intracellular Ca2+ release from ryanodine-sensitive stores66); 2) α1A-AR-mediated, phenylephrine-induced contractions in rat tail and iliac arteries involved VDCC-dependent Ca2+ influxes, the activation of which was triggered by cation influxes sensitive to LOE 908, an inhibitor of receptor-operated Ca2+ channels (ROCCs)67); 3) α1B-AR-mediated, phenylephrine-induced contractions in rat spleen involved Ca2+ influxes sensitive to SKF-96365, an inhibitor of ROCC and store-operated Ca2+ channels (SOCCs), and intracellular Ca2+ release68); and 4) α1D-AR-mediated, NA-induced contractions in the rat aorta involved nifedipine-sensitive and SKF-96365-sensitive Ca2+ influxes.69) These findings suggest that VDCC-independent and -dependent Ca2+ entry routes trigger α1H-AR-mediated SM contractions. However, to the best of our knowledge, the dependency on extracellular Ca2+ and its entry routes responsible for α1L-AR-mediated SM contractions have not been studied to date.
In this study, to clarify the extracellular Ca2+ entry routes responsible for α1L-AR-mediated SM contractions, we focused on the guinea pig (GP) thoracic aorta (TA), in which NA is reported to induce α1L-AR-mediated contractions.60) GP TA is a commonly used tissue for pharmacological studies and could be more suitable for studying α1L-AR than resistant arteries and lower urinary tract tissues, in which α1H-AR subtypes may be present in addition to α1L-AR (Table 1). Here, we report that α1L-AR-mediated, phenylephrine-induced contractions in the GP TA are triggered by Ca2+ influxes through both SOCCs and VDCCs. We also suggest that the principal SOCC candidates are ORAI3/ORAI1 and stromal interaction molecule 2 (STIM2) based on their mRNA expression in GP TA.
MATERIALS AND METHODS
AnimalsThis study was approved by the Toho University Animal Care and User Committee (Approval Nos. 20-51-444, accredited on May 19, 2020; 21-52-444, accredited on April 21, 2021; 22-53-444, accredited on April 9, 2022). The experiments were conducted in accordance with the guidelines of the Laboratory Animal Center of the Faculty of Pharmaceutical Sciences, Toho University. For this study, we purchased male GPs (age, 5–11 weeks; weight, 287–550 g) from Kyudo, Co., Ltd. (Saga, Japan). The GPs were housed under controlled conditions (21–22 °C, relative air humidity 50 ± 5%) and a fixed 12/12 h light–dark cycle (08:00–20:00), with food and water available ad libitum.
Preparation of GP TAThe GPs were anesthetized using isoflurane inhalation and euthanized by exsanguination via the carotid artery. The isolated TAs were cleaned of connective tissue and fat and cut into ring preparations (approximately 2 mm in length) in normal Tyrode’s solution containing (mM): NaCl, 158.3; KCl, 4.0; CaCl2, 2.0; MgCl2, 1.05; NaH2PO4, 0.42; NaHCO3, 10.0; and D-(+)-glucose, 5.6. The intimal surface of ring preparations was rubbed to remove the endothelium.
Measurement of Tension ChangesThe ring preparations were suspended under an optimal resting tension of 1.0 g using stainless steel hooks (outer diameter, 200 µm) in a 5-mL organ bath (UC-5, UFER Medical Instrument, Kyoto, Japan) containing normal Tyrode’s solution. The resting tension was determined according to methods previously described.60) The solution was maintained at 35.0 ± 1.0 °C (pH = 7.4) and continuously bubbled with a mixture of 95% O2 and 5% CO2. TA tension changes were isometrically recorded using force-displacement transducers (T7-8-240, Orientec, Tokyo, Japan; ULA-10GR, MinebeaMitsumi Inc., Nagano, Japan) connected to amplifiers (AM20AZ, Unipulse Corporation, Tokyo, Japan; AP-621G, Nihon Kohden, Tokyo, Japan) and PowerLab™/LabChart™ software (ADInstruments Pty., Ltd., Bella Vista, NSW, Australia). After equilibrated for 60 min in normal Tyrode’s solution, the TA segments were contracted with phenylephrine (10−5 M). After then, to verify the functional absence of endothelium, the TA segments were challenged with acetylcholine (ACh, 10−5 M). TA preparations hardly affected by ACh were regarded as endothelium-denuded TA preparations. The TA preparations were then contracted four times with phenylephrine (3 × 10−4 M). To prevent any possible effects of endogenous prostaglandins, all experiments were performed in the presence of indomethacin (3 × 10−6 M).
Effects of α-AR Antagonists and Ca2+ Channel Inhibitors on Concentration–Response Curves (CRCs) for PhenylephrineAfter the procedures described in “Measurement of Tension Changes,” to obtain a CRC for the control, phenylephrine was cumulatively added to the bath solution. For some experiments, the vehicle (0.2% ethanol (EtOH) for silodosin; 6 × 10−4, 0.02, and 0.3% dimethyl sulfoxide (DMSO) for L-765314, YM-254890, and LOE 908, respectively), and/or verapamil (a VDCC inhibitor, 10−5 M) was administered 20 min before control CRC determination. After washing out, prazosin (an α1-AR antagonist, 3 × 10−9–3 × 10−8 M), tamsulosin (an α1A-AR antagonist, 3 × 10−10–3 × 10−9 M), silodosin (an α1A-AR antagonist, 10−9–3 × 10−8 M), L-765314 (an α1B-AR antagonist, 3 × 10−8 M), BMY 7378 (an α1D-AR antagonist, 10−8 M), yohimbine (an α2-AR antagonist, 10−8 M), idazoxan (an α2-AR antagonist, 10−8 M), YM-254890 (a Gq protein inhibitor, 10−6 M), verapamil (10−5 M), verapamil (10−5 M) + LOE 908 (3 × 10−5 M), or verapamil (10−5 M) + SKF-96365 (3 × 10−5 M) was applied in the bath medium and incubated for 20 min. The concentrations and incubation time of the antagonists and inhibitors used in this study were sufficient to inhibit the targeted receptors and channels based on previous reports.20,43–46,60,70–75) After then, to obtain the CRC in the presence of drugs, phenylephrine was cumulatively added again.
To examine the effects of the Ca2+-free solution, the bath solution was replaced 20 min before CRC with the following solution (mM): NaCl, 158.3; KCl, 4.0; MgCl2, 1.05; NaH2PO4, 0.42; NaHCO3, 10.0; D-(+)-glucose, 5.6; and ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N'-tetraacetic acid (EGTA), 0.2.
RT-Quantitative (q)PCR of α-AR, SOCC-Related, and ROCC-Related mRNA ExpressionIn the mRNA experiment, the endothelium of the TA preparations was carefully denuded. RT-qPCR was performed according to the methods described in a previous report.76) Briefly, total RNA was extracted from the endothelium-denuded TA preparations by the acid guanidinium thiocyanate-phenol-chloroform method. After treated with deoxyribonuclease (Nippon Gene Co., Ltd., Tokyo, Japan) for 30 min at 37 °C, total RNA pellets were obtained by phenol-chloroform extraction/ethanol precipitation and dissolved in diethyl pyrocarbonate-treated water. We used ReverTra Ace® qPCR RT Master Mix with gDNA Remover (TOYOBO Co., Ltd., Osaka, Japan) to synthesize first-strand cDNA by reverse transcription. RT-qPCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Waltham, MA, U.S.A.) using THUNDERBIRD® Next SYBR® qPCR Mix (TOYOBO Co., Ltd.). The primers used in this study are shown in Supplementary Table 1. The thermal cycler parameters were set to 95 °C for 2 min, followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 35 s. Fluorescence intensity was measured at each 72 °C step. The mRNA expression level of each gene was analyzed by Sequence Detection Software (Applied Biosystems) and calculated as a relative value to that of β-actin gene (Actb), which was set to 1. We considered the mRNA expression level as 0 for samples that did not reach the fluorescence intensity threshold after 40 cycles.
DrugsThe following drugs were used in this study: l-phenylephrine hydrochloride, idazoxan hydrochloride, (±)-verapamil hydrochloride, and indomethacin (Sigma-Aldrich Co., St. Louis, MO, U.S.A.); prazosin hydrochloride, silodosin, yohimbine hydrochloride, and YM-254890 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan); tamsulosin hydrochloride (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan); L-765314 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, U.S.A.); BMY 7378 dihydrochloride (Research Biochemicals Inc., MA, U.S.A.); LOE 908 (Nippon Boehringer Ingelheim Co., Ltd., Hyogo, Japan); SKF-96365 (Cayman Chemical Co., Ann Arbor, MI, U.S.A. or Tokyo Chemical Industry Co., Ltd.); and Ach chloride (Daiichi Sankyo Co., Ltd., Tokyo, Japan).
Silodosin was dissolved in EtOH to prepare stock solutions of 5 × 10−3 M and diluted with EtOH to the desired concentrations. Indomethacin was dissolved in EtOH to prepare a stock solution of 10−2 M. L-765314/YM-254890 and LOE 908 were dissolved in DMSO to prepare stock solutions of 5 × 10−3 M and 10−2 M. All the other drugs were dissolved/diluted with distilled water.
Data and Statistical AnalysesThe extent of phenylephrine-induced contractions was calculated relative to the tension level before the phenylephrine administration (0% contraction) and to the 3 × 10−4 M phenylephrine-induced contractions in the control CRC (100% contraction). The data were plotted as a function of phenylephrine concentration and fitted to the following equation:
where E is % contraction at a given phenylephrine concentration, Emax is the maximum contraction, A is the phenylephrine concentration, nH is the Hill coefficient, and EC50 is the phenylephrine concentration that produces a half-maximal response. Curve fitting was performed using GraphPad Prism™ (GraphPad Software Inc., San Diego, CA, U.S.A.). α-AR antagonist potencies were expressed as pA2 values, which were calculated from a Schild plot analysis.77)
Data are expressed as the mean ± standard error of the mean (S.E.M.) or the mean with 95% confidence interval (95% CI), where n refers to the number of experiments. Statistical analyses were performed by Šidák’s test after two-way ANOVA using GraphPad Prism™. All statistical analyses were conducted with a significance level of α = 0.05 (p < 0.05).
RESULTS
Effects of Prazosin, Tamsulosin, and Silodosin on Phenylephrine-Induced ContractionsPrazosin (3 × 10−9–3 × 10−8 M) inhibited the phenylephrine-induced contractions and shifted the CRCs for phenylephrine to the right (Fig. 1Aa). The slope of the regression line for the Schild plot of prazosin vs. phenylephrine was 1.06 (95% CI: 0.85–1.27), which was not significantly different from unity (Fig. 1Ab). The pA2 value of prazosin vs. phenylephrine was 8.53 (95% CI: 8.42–8.69).
Tamsulosin (3 × 10−10–3 × 10−9 M) inhibited the phenylephrine-induced contractions and shifted the CRCs for phenylephrine to the right (Fig. 1Ba). The slope of the regression line for the Schild plot of tamsulosin vs. phenylephrine was 1.14 (95% CI: 0.88–1.41), which was not significantly different from unity (Fig. 1Bb). The pA2 value of tamsulosin vs. phenylephrine was 9.74 (95% CI: 9.58–9.99).
Silodosin (10−9–3 × 10−8 M) inhibited the phenylephrine-induced contractions and shifted the CRCs for phenylephrine to the right (Fig. 1Ca). However, the slope of the regression line for the Schild plot of silodosin vs. phenylephrine was 0.58 (95% CI: 0.37–0.78), which was significantly less than unity (Fig. 1Cb). The apparent pA2 value of silodosin vs. phenylephrine was 9.48 (95% CI: 9.12–10.19).
Effects of L-765314, BMY 7378, Yohimbine, and Idazoxan on Phenylephrine-Induced ContractionsNeither L-765314 (3 × 10−8 M, Fig. 2A) nor BMY 7378 (10−8 M, Fig. 2B) significantly affected the phenylephrine-induced contractions (vs. DMSO/control).
Both yohimbine (10−8 M, Fig. 2C) and idazoxan (10−8 M, Fig. 2D) significantly, but only slightly, inhibited or augmented the phenylephrine-induced contractions (vs. control). We concluded that the effects of yohimbine and idazoxan were negligible.
Comparison of mRNA Expression Levels of α-ARs Assessed by RT-qPCRThe relative mRNA expression levels of α-ARs (α1A-AR (Adra1a), α1B-AR (Adra1b), α1D-AR (Adra1d), α2A-AR (Adra2a), α2B-AR (Adra2b), and α2C-AR (Adra2c)) in endothelium-denuded TA are shown in Fig. 3. The most abundant α-AR mRNA was Adra1a, followed by Adra1d, Adra1b, and Adra2c. In contrast, mRNA expression levels of Adra2a and Adra2b were the lowest.
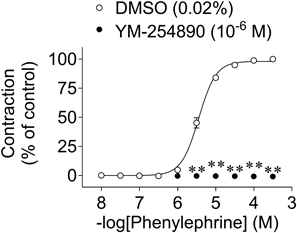
Effects of YM-254890 on Phenylephrine-Induced ContractionsYM-254890 (10−6 M) completely inhibited the phenylephrine-induced contractions (Fig. 4).
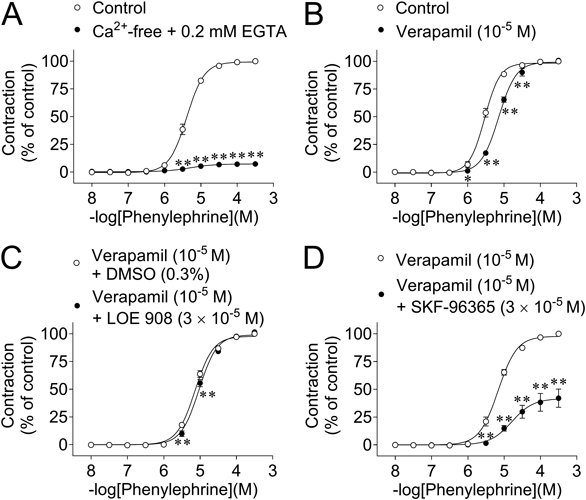
Effects of Verapamil and Removal of Ca2+ on Phenylephrine-Induced ContractionsThe CRCs for phenylephrine-induced contractions in normal Tyrode’s solution and Ca2+-free solution containing 0.2 mM EGTA are shown in Fig. 5A. The phenylephrine-induced contractions were strongly inhibited by the Ca2+-free solution; at 3 × 10−4 M phenylephrine, the contraction was inhibited by approx. 95% (Fig. 5A).
Verapamil (10−5 M) significantly inhibited the contractions at low concentrations (10−6–3 × 10−5 M) (vs. control) (Fig. 5B). In contrast, the contractions were not affected by verapamil (10−5 M) at higher concentrations (≥10−4 M).
Effects of LOE 908 and SKF-96365 on Phenylephrine-Induced Contractions in the Presence of VerapamilLOE 908 (3 × 10−5 M) slightly but significantly inhibited the CRCs for phenylephrine-induced contractions in the presence of verapamil (10−5 M) (vs. verapamil + DMSO) (Fig. 5C).
SKF-96365 (3 × 10−5 M) strongly inhibited the CRCs for phenylephrine-induced contractions in the presence of verapamil (10−5 M); at 3 × 10−4 M, the contraction was inhibited by approx. 60% (Fig. 5D).
Comparison of mRNA Expression Levels of SOCC-Related Molecules (Orai and Stim) Assessed by RT-qPCRThe relative mRNA expression levels of SOCC-related molecules (Orai (Orai1–3) and Stim (Stim1 and Stim2)) in endothelium-denuded TA are shown in Fig. 6. The most abundant SOCC-related molecule mRNA was Orai3 and Stim2, followed by Orai1. In contrast, the mRNA expression levels of Orai2 and Stim1 were the lowest.
Comparison of mRNA Expression Levels of ROCC-Related Molecules (Transient Receptor Potential (TRP) Channels) Assessed by RT-qPCRThe relative mRNA expression levels of ROCC-related molecules (TRP channels (Trpc1, Trpc3, Trpc4, Trpc5, Trpc6, Trpc7, Trpm8, and Trpv4)) in endothelium-denuded TA are shown in Supplementary Fig. 1. We tested the presence of TRPC channels which are considered to play a major role in receptor-operated Ca2+ entry in SMs78) and TRPM8/TRPV4 channels which are reported to involve rat aortic SM contractility.79) The most abundant TRP channel mRNA was Trpc6, followed by Trpc1 and Trpc3. The mRNA expression levels of the other TRP channels were very low compared with the Trpc6 mRNA level, in the following order of expression: Trpc5 ≈ Trpv4 > Trpc4 ≈ Trpc7 ≫ Trpm8.
DISCUSSION
We showed that phenylephrine-induced contractions in GP TA SMs were triggered by the stimulation of Gq protein-coupled α1L-AR, which was followed by Ca2+ influx through SOCCs (ORAI1/3 and STIM2) and VDCCs. GP TA could be more suitable for studying α1L-AR than resistant arteries and lower urinary tract tissues, in which α1H-AR subtypes may be present in addition to α1L-AR (Table 1). Furthermore, its easy-to-handle properties will contribute to the progress of α1L-AR research.
First, we discuss the effect of prazosin on phenylephrine-induced contractions in GP TA. These contractions were competitively suppressed by prazosin, and its pA2 value was calculated to be 8.53 (Fig. 1A). Since this pA2 value was <9, we concluded that the α1-AR responsible for phenylephrine-induced contractions was α1L-AR, as α1H-AR has a pA2 > 9.43–46) Yamamoto and Koike also reported that the α1-AR mediating NA-induced contractions in GP TA was α1L-AR.60)
Second, we discuss the effects of α1A-AR antagonists (tamsulosin/silodosin) on phenylephrine-induced contractions. α1L-AR is defined as α1-AR showing low affinity for prazosin and high affinity for α1A-AR antagonists such as tamsulosin/silodosin (pA2 ≈ 10).43–46) In the present study, the phenylephrine-induced contractions of GP TA were competitively suppressed by tamsulosin, with a pA2 value of 9.74 (Fig. 1B). This value is consistent with the pA2 or pKB values of tamsulosin for α1L-AR: 9.57 (pA2 vs. NA/GP TA)60) and 9.99 (pKB vs. phenylephrine/rabbit prostate).80)
We also tested the effects of silodosin, another specific inhibitor of α1A-AR. Silodosin is reported to show a competitive antagonistic action on α1A-AR/α1L-AR in the following SM tissues based on the regression line slopes of the Schild plots for silodosin vs. the α1-AR agonists being unity: hamster ureter (pA2 = 9.44 vs. phenylephrine), rat tail artery (pA2 = 10.0 vs. NS-49 (an α1A-AR agonist)), dog mesenteric artery (pA2 = 9.9 vs. NS-49), and rabbit ureter (pA2 = 8.71 vs. NA).24,81,82) Although the phenylephrine-induced contractions were suppressed by silodosin in a concentration-dependent manner (Fig. 1Ca), the slope of the regression line for the Schild plot of silodosin vs. phenylephrine was less than unity (0.58 (95% CI: 0.37–0.78)) (Fig. 1Cb), which does not support a competitive antagonist action vs. phenylephrine. Similar results have been reported in human and rabbit prostates, in which α1L-AR mediates NA-/phenylephrine-induced contractions. In both SM tissues, Schild plot analysis was not performed for silodosin-induced inhibition, possibly because of its lack of apparent competitive antagonistic action. In these studies, instead of pA2 values, apparent pKB values were calculated for silodosin (9.45 vs. NA, human prostate; 10.05/9.6 vs. phenylephrine/NA, rabbit prostate).80,83,84) Furthermore, in human prostate tissue, silodosin was suggested to be prone to bind to non-α1-AR sites as well as α1-AR. Namely, the specific binding of [3H]silodosin (500 pM) was shown to be almost equal to the non-specific binding of [3H]silodosin (500 pM).85) Therefore, in GP TA and in prostate tissues, the apparent non-competitive antagonistic action of silodosin vs. phenylephrine might be partly explained by its tendency to bind to non-α1-AR sites. Although competitive antagonism of silodosin vs. phenylephrine was not observed in GP TA, its apparent pA2 value was tentatively calculated to be 9.48, which is consistent with the values for α1A-AR/α1L-AR. Therefore, we conclude that the pharmacological characteristics of α1L-AR, which mediates phenylephrine-induced contractions in GP TA, are almost the same as those in other SM tissues.
Third, we discuss the effects of other α-AR (α1B-, α1D-, and α2-AR) antagonists on the phenylephrine-induced contractions. These contractions were not substantially suppressed by L-765314 (an α1B-AR antagonist, 3 × 10−8 M), BMY 7378 (an α1D-AR antagonist, 10−8 M), yohimbine (an α2-AR antagonist, 10−8 M), and idazoxan (an α2-AR antagonist, 10−8 M) (Fig. 2). Therefore, α1B-AR/α1D-AR/α2-AR are unlikely to be involved in the phenylephrine-induced contractions and this is supported for the following reasons: 1) the concentration of L-765314 used in this study (30 nM) could sufficiently antagonize α1B-AR since its Ki value for α1B-AR is 5.4 nM70); 2) the concentration of BMY 7378 used in this study (10 nM) could sufficiently antagonize α1D-AR since its Ki value for α1D-AR is 0.43 nM20); and 3) the concentrations of yohimbine and idazoxan used in this study (10−8 M, negative logarithm of 8.0) could sufficiently antagonize α2-AR since the pA2 values of yohimbine and idazoxan for α2-AR calculated using rat vas deferens are reported to be 8.24 and 8.70, respectively.71)
The pharmacological characteristics of the α-AR responsible for phenylephrine-induced contractions in GP TA are consistent with those of α1L-AR, which is proposed to be derived from the α1A-AR mRNA (Adra1a).43–46) This is also supported by our present mRNA expression results, showing that Adra1a is the most abundant α-AR (Fig. 3).
Sex differences in α-AR have been reported as follows: 1) Administration of phenylephrine dose-dependently reduced finger blood flow in men, but not in women.86) 2) The number of α-ARs in platelets of women who are hypertensive is 1.5 times higher than in those of women who are normotensive, but the affinity of [3H]dihydroergocryptine for α-AR did not differ between these two groups, or between men and women.87) 3) The binding properties of [3H]prazosin to the rabbit urethra are the same in males and females.88) In this study, we determined that α1L-AR is predominant in GP TA using only male GPs. Further studies using female GPs are required to clarify whether there are sex differences in the function and subtype of α1-ARs in the GP TA.
The pathophysiological role of α1L-AR in GP TA is currently unclear. However, in rat femoral arteries in which α1H-AR and α1L-AR are present, the α1-AR subtypes mediating contraction in the spontaneously hypertensive rat femoral artery were identical with those in the normotensive rat tissue.63) In addition, a study using young adult and aged rat urinary bladder SM suggests that the promotion of contractile responses mediated by α1A-AR, but not α1L-AR, may be the cause of unstable bladder in those who are elderly.19) Therefore, α1L-AR is unlikely to actively be involved in hypertension and unstable bladder in older individuals. Moreover, in human benign prostatic hyperplasia tissue in which α1H-AR and α1L-AR are present, treatment with α1-AR antagonists may not change the conditions of α1-ARs in this tissue.89) Thus, α1-AR antagonists that strongly suppress α1L-AR are needed for the treatment of benign prostatic hyperplasia.
Fourth, we discuss the post-receptor mechanisms of phenylephrine-induced contractions. These contractions were completely suppressed by a Gq-selective inhibitor YM-25489090) (Fig. 4), suggesting that they were mediated by Gq protein activation. In addition, the phenylephrine-induced contractions were potently suppressed by the removal of extracellular Ca2+ (Fig. 5A). This finding indicates that these contractions strongly depend on extracellular Ca2+ influx. One extracellular Ca2+ influx route was indicated to be VDCC-mediated because verapamil (a VDCC inhibitor) partly but significantly suppressed the contractions induced by low concentrations (10−6–3 × 10−5 M) of phenylephrine (vs. control) (Fig. 5B). The concentration of verapamil (10−5 M) used in this study was sufficient to inhibit VDCC because it completely suppressed the binding of [3H]D888 (4.2 nM) to rat cardiac membranes in a previous study.72) Therefore, VDCC plays an important role in the Ca2+ influx pathway when α1L-AR is weakly activated. However, VDCC is not the only Ca2+ influx pathway, and the contribution of VDCC-independent Ca2+ influx is speculated to be substantial because the contractions induced by high concentrations of phenylephrine (≥10−4 M) were not affected by verapamil (10−5 M) (Fig. 5B). VDCC-independent Ca2+ influx is the main pathway when α1L-AR is strongly activated. In support of our results, Tanaka et al. also reported that the contractile responses of GP TA induced by a high concentration (3 × 10−5 M) of NA were not suppressed by various VDCC inhibitors (nicardipine/diltiazem/verapamil).73) In the mouse prostate, a VDCC inhibitor (nifedipine) has been reported to suppress α1L-AR-mediated contractions induced by electrical field stimulation, but the suppression rate differs depending on the frequency of electrical stimulation.91)
To clarify the possible involvement of ROCCs and SOCCs in VDCC-independent Ca2+ influx pathways, the effects of LOE 908 and SKF-96365 on the phenylephrine-induced contractions were investigated. LOE 908 inhibits ROCCs, although it also inhibits VDCCs.92) SKF-96365 is an inhibitor of SOCCs/ROCCs and shows inhibitory effects on VDCCs.92,93) Therefore, in this study, the inhibitory effects of LOE 908 for ROCC were evaluated in the presence of verapamil, and those of SKF-96365 for SOCC were evaluated in the presence of verapamil but in the absence of LOE 908 since the effects of LOE 908 were very limited. Our findings showed that the inhibitory effects of LOE 908 (3 × 10−5 M) vs. phenylephrine were significant, but only slight (Fig. 5C), and those of SKF-96365 (3 × 10−5 M) were very strong (Fig. 5D). Therefore, SOCCs are suggested to represent the principal VDCC-independent Ca2+ influx pathway, although ROCCs may be involved. In GP TA, SKF-96365-inhibitable characteristics have also been reported for NA-induced contractions.73) An important role of SOCCs in α1-AR-mediated contraction has also been reported in non-vascular SM tissues. Specifically, in mouse urethral SM, phenylephrine-induced contractions were strongly attenuated by SOCC inhibitors,94) although it is unclear whether α1A-AR/α1L-AR is predominant in this tissue.7) To the best of our knowledge, our study is the first to show that α1L-AR-mediated SM contraction is triggered by SOCCs/VDCCs through Gq protein activation. However, to clarify whether functional coupling of α1L-AR with SOCCs/VDCCs is conserved among SM type, further studies are required using tissues in which α1L-AR functionally predominates, such as genital and lower urinary tract SMs.
Finally, we discuss the molecular candidates for SOCC/ROCC that are activated by the stimulation of α1L-AR. ORAI channels are molecular candidates for the SOCC, and ORAI channel activation is regulated by STIMs, which are Ca2+ sensors of the endoplasmic reticulum.95) To identify the SOCC/STIM molecular candidates responsible for phenylephrine-induced α1L-AR-mediated contractions, the Orai and Stim mRNA expression levels were assessed by RT-qPCR. Among the tested Orai and Stim mRNA, Orai3 and Stim2 mRNA expression levels were the highest in GP TA, followed by Orai1 (Fig. 6). Therefore, stimulation of α1L-AR in the GP TA is speculated to activate ORAI1 and/or ORAI3 through STIM2. In addition, TRP channels are molecular candidates for the ROCC.96) The relative mRNA expression of the tested TRP channels in GP TA was Trpc6 > Trpc1 > Trpc3 (Supplementary Fig. 1). Therefore, in addition to SOCCs, these TRP channels may function as VDCC-independent Ca2+ influx pathways in α1L-AR-mediated GP TA contractions, although their contributions are very small compared to those of SOCCs/VDCCs. Another role of ROCCs in SM contraction is to trigger the activation of VDCCs.97) Therefore, the verapamil-sensitive component of the phenylephrine-induced α1L-AR-mediated contractions may be generated by being triggered by LOE 908-sensitive ROCCs. By using ROCC inhibitors without VDCC inhibition, this role of ROCCs may be clarified, and this issue should be studied in the future.
Acknowledgments
The authors would like to thank Dr. Fumiko Yamaki for her expert technical assistance.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Zhong H, Minneman KP. Alpha1-adrenoceptor subtypes. Eur. J. Pharmacol., 375, 261–276 (1999).
- 2) Langer SZ. Presynaptic regulation of the release of catecholamines. Pharmacol. Rev., 32, 337–362 (1980).
- 3) Furukawa K, Rosario DJ, Smith DJ, Chapple CR, Uchiyama T, Chess-Williams R. Alpha 1A-adrenoceptor-mediated contractile responses of the human vas deferens. Br. J. Pharmacol., 116, 1605–1610 (1995).
- 4) Moriyama N, Nasu K, Takeuchi T, Akiyama K, Murata S, Nishimatsu H, Yano J, Tsujimoto G, Kawabe K. Quantification and distribution of alpha 1-adrenoceptor subtype mRNAs in human vas deferens: comparison with those of epididymal and pelvic portions. Br. J. Pharmacol., 122, 1009–1014 (1997).
- 5) Marshall I, Burt RP, Chapple CR. Noradrenaline contractions of human prostate mediated by alpha 1A-(alpha 1c-) adrenoceptor subtype. Br. J. Pharmacol., 115, 781–786 (1995).
- 6) El-Gamal OM, Sandhu DP, Terry T, Elliott RA. alpha-Adrenoceptor subtypes in isolated corporal tissue from patients undergoing gender re-assignment. BJU Int., 97, 329–332 (2006).
- 7) Alexandre EC, de Oliveira MG, Campos R, Kiguti LR, Calmasini FB, Silva FH, Grant AD, Yoshimura N, Antunes E. How important is the α1-adrenoceptor in primate and rodent proximal urethra? Sex differences in the contribution of α1-adrenoceptor to urethral contractility. Am. J. Physiol. Renal Physiol., 312, F1026–F1034 (2017).
- 8) Yano S, Hirose M, Nakada T, Nakayama J, Matsuo K, Yamada M. Selective alpha 1A-adrenoceptor stimulation induces Mueller’s smooth muscle contraction in an isolated canine upper eyelid preparation. Curr. Eye Res., 35, 363–369 (2010).
- 9) Daniel EE, Brown RD, Wang YF, Low AM, Lu-Chao H, Kwan CY. Alpha-adrenoceptors in canine mesenteric artery are predominantly 1A subtype: pharmacological and immunochemical evidence. J. Pharmacol. Exp. Ther., 291, 671–679 (1999).
- 10) Alberts P, Bergström PA, Fredrickson MG. Characterisation of the functional alpha-adrenoceptor subtype in the isolated female pig urethra. Eur. J. Pharmacol., 371, 31–38 (1999).
- 11) Yan M, Zhang Y, Du XJ, Han C. Functional assessment of alpha 1-adrenoceptor subtypes in porcine coronary artery. Clin. Exp. Pharmacol. Physiol., 25, 682–685 (1998).
- 12) Takayanagi I, Harada M, Koike K, Satoh M. Differences in alpha 1-adrenoceptor mechanisms for phenylephrine and tizanidine in rabbit thoracic aorta and common iliac artery. Can. J. Physiol. Pharmacol., 69, 1819–1824 (1991).
- 13) Takayanagi I, Moriya M. Alpha 1A-adrenoceptors in rabbit bronchus. Eur. J. Pharmacol., 202, 281–283 (1991).
- 14) Aboud R, Shafii M, Docherty JR. Investigation of the subtypes of alpha 1-adrenoceptor mediating contractions of rat aorta, vas deferens and spleen. Br. J. Pharmacol., 109, 80–87 (1993).
- 15) Burt RP, Chapple CR, Marshall I. Evidence for a functional alpha 1A- (alpha 1C-) adrenoceptor mediating contraction of the rat epididymal vas deferens and an α1B-adrenoceptor mediating contraction of the rat spleen. Br. J. Pharmacol., 115, 467–475 (1995).
- 16) Campos M, de Lucena Morais P, Pupo AS. Functional characterisation of alpha(1)-adrenoceptors in denervated rat vas deferens. Naunyn Schmiedebergs Arch. Pharmacol., 368, 72–78 (2003).
- 17) Bültmann R, Kurz AK, Starke K. Alpha 1-adrenoceptors and calcium sources in adrenergic neurogenic contractions of rat vas deferens. Br. J. Pharmacol., 111, 151–158 (1994).
- 18) Couldwell C, Jackson A, O’Brien H, Chess-Williams R. Characterization of the alpha1-adrenoceptors of the rat prostate gland. J. Pharm. Pharmacol., 45, 922–924 (1993).
- 19) Suzuki Y, Moriyama N, Kanada A, Okaya Y, Kawabe K, Aisaka K. The role of alpha 1L-adrenoceptor in rat urinary bladder: comparison between young adult and aged rats. Life Sci., 65, 2553–2559 (1999).
- 20) Chess-Williams R, Aston N, Couldwell C. Alpha 1A-adrenoceptor subtype mediates contraction of the rat urethra. J. Auton. Pharmacol., 14, 375–381 (1994).
- 21) Pacini ESA, Castilho ACS, Hebeler-Barbosa F, Pupo AS, Kiguti LRA. Contraction of rat cauda epididymis smooth muscle to α1-adrenoceptor activation is mediated by α1A-adrenoceptors. J. Pharmacol. Exp. Ther., 366, 21–28 (2018).
- 22) Piascik MT, Hrometz SL, Edelmann SE, Guarino RD, Hadley RW, Brown RD. Immunocytochemical localization of the alpha-1B adrenergic receptor and the contribution of this and the other subtypes to vascular smooth muscle contraction: analysis with selective ligands and antisense oligonucleotides. J. Pharmacol. Exp. Ther., 283, 854–868 (1997).
- 23) Chen H, Bischoff A, Schäfers RF, Wambach G, Philipp T, Michel MC. Vasoconstriction of rat renal interlobar arteries by noradrenaline and neuropeptide Y. J. Auton. Pharmacol., 17, 137–146 (1997).
- 24) Tomiyama Y, Kobayashi K, Tadachi M, Kobayashi S, Inada Y, Kobayashi M, Yamazaki Y. Expressions and mechanical functions of alpha1-adrenoceptor subtypes in hamster ureter. Eur. J. Pharmacol., 573, 201–205 (2007).
- 25) Hatano A, Takahashi H, Tamaki M, Komeyama T, Koizumi T, Takeda M. Pharmacological evidence of distinct alpha 1-adrenoceptor subtypes mediating the contraction of human prostatic urethra and peripheral artery. Br. J. Pharmacol., 113, 723–728 (1994).
- 26) Giessler C, Wangemann T, Silber RE, Dhein S, Brodde OE. Noradrenaline-induced contraction of human saphenous vein and human internal mammary artery: involvement of different alpha1-adrenoceptor subtypes. Naunyn Schmiedebergs Arch. Pharmacol., 366, 104–109 (2002).
- 27) Furukawa K, Chess-Williams R, Uchiyama T. Alpha 1B-adrenoceptor subtype mediating the phenylephrine-induced contractile response in rabbit corpus cavernosum penis. Jpn. J. Pharmacol., 71, 325–331 (1996).
- 28) Smith KM, Macmillan JB, McGrath JC. Investigation of alpha1-adrenoceptor subtypes mediating vasoconstriction in rabbit cutaneous resistance arteries. Br. J. Pharmacol., 122, 825–832 (1997).
- 29) Eltze M. Characterization of the alpha 1-adrenoceptor subtype mediating contraction of guinea-pig spleen. Eur. J. Pharmacol., 260, 211–220 (1994).
- 30) Jähnichen S, Eltze M, Pertz HH. Evidence that alpha(1B)-adrenoceptors are involved in noradrenaline-induced contractions of rat tail artery. Eur. J. Pharmacol., 488, 157–167 (2004).
- 31) Villalobos-Molina R, Ibarra M. Alpha 1-adrenoceptors mediating contraction in arteries of normotensive and spontaneously hypertensive rats are of the alpha 1D or alpha 1A subtypes. Eur. J. Pharmacol., 298, 257–263 (1996).
- 32) Sayet I, Neuilly G, Rakotoarisoa L, Mironneau C, Mironneau J. Relation between alpha 1-adrenoceptor subtypes and noradrenaline-induced contraction in rat portal vein smooth muscle. Br. J. Pharmacol., 110, 207–212 (1993).
- 33) Eltze M. Functional evidence for an alpha 1B-adrenoceptor mediating contraction of the mouse spleen. Eur. J. Pharmacol., 311, 187–198 (1996).
- 34) Satoh M, Enomoto K, Takayanagi I, Koike K. Analysis of alpha1-adrenoceptor subtypes in rabbit aorta and arteries: regional difference and co-existence. Eur. J. Pharmacol., 374, 229–240 (1999).
- 35) Testa R, Destefani C, Guarneri L, Poggesi E, Simonazzi I, Taddei C, Leonardi A. The alpha 1d-adrenoceptor subtype is involved in the noradrenaline-induced contractions of rat aorta. Life Sci., 57, PL159–PL163 (1995).
- 36) Asbún-Bojalil J, Castillo EF, Escalante BA, Castillo C. Does segmental difference in alpha 1-adrenoceptor subtype explain contractile difference in rat abdominal and thoracic aortae? Vascul. Pharmacol., 38, 169–175 (2002).
- 37) Hussain MB, Marshall I. Characterization of alpha1-adrenoceptor subtypes mediating contractions to phenylephrine in rat thoracic aorta, mesenteric artery and pulmonary artery. Br. J. Pharmacol., 122, 849–858 (1997).
- 38) Nomura S, Sunagane N, Uruno T, Kubota K. Further characterization of antagonism by alpha 2-adrenoceptor agonists of contractions induced by alpha 1-adrenoceptor agonists. Res. Commun. Mol. Pathol. Pharmacol., 92, 31–42 (1996).
- 39) Yousif M, Kadavil EA, Oriowo MA. Heterogeneity of alpha 1-adrenoceptor subtypes mediating noradrenaline-induced contractions of the rat superior mesenteric artery. Pharmacology, 56, 196–206 (1998).
- 40) Piascik MT, Guarino RD, Smith MS, Soltis EE, Saussy DL Jr, Perez DM. The specific contribution of the novel alpha-1D adrenoceptor to the contraction of vascular smooth muscle. J. Pharmacol. Exp. Ther., 275, 1583–1589 (1995).
- 41) Bexis S, Cleary L, McGrath JC, Tanoue A, Tsujimoto G, Docherty JR. Alpha(1D)-adrenoceptors mediate nerve and agonist-evoked contractions in mouse vas deferens: evidence obtained from knockout technology. Auton. Autacoid Pharmacol., 28, 81–85 (2008).
- 42) Yamamoto Y, Koike K. Characterization of alpha1-adrenoceptor-mediated contraction in the mouse thoracic aorta. Eur. J. Pharmacol., 424, 131–140 (2001).
- 43) Docherty JR. Subtypes of functional alpha1-adrenoceptor. Cell. Mol. Life Sci., 67, 405–417 (2010).
- 44) White CW, da Silva Junior ED Junior, Lim L, Ventura S. What makes the α1A-adrenoceptor gene product assume an α1L-adrenoceptor phenotype? Br. J. Pharmacol., 176, 2358–2365 (2019).
- 45) Muramatsu I, Suzuki F, Tanaka T, Yamamoto H, Morishima S. Alpha1-adrenoceptor subtypes and alpha1-adrenoceptor antagonists. Yakugaku Zasshi, 126, 187–198 (2006).
- 46) Nishimune A, Yoshiki H, Uwada J, Anisuzzaman AS, Umada H, Muramatsu I. Phenotype pharmacology of lower urinary tract α(1)-adrenoceptors. Br. J. Pharmacol., 165, 1226–1234 (2012).
- 47) Ford AP, Arredondo NF, Blue DR Jr, Bonhaus DW, Jasper J, Kava MS, Lesnick J, Pfister JR, Shieh IA, Vimont RL, Williams TJ, McNeal JE, Stamey TA, Clarke DE. RS-17053 (N-[2-(2-cyclopropylmethoxyphenoxy)ethyl]-5-chloro-alpha, alpha-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective alpha 1A-adrenoceptor antagonist, displays low affinity for functional alpha 1-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol. Pharmacol., 49, 209–215 (1996).
- 48) Davis BJ, Wiener M, Chapple CR, Sellers DJ, Chess-Williams R. Functional and radioligand binding characterization of the α1L-adrenoceptor subtype of the human vas deferens. Auton. Autacoid Pharmacol., 34, 41–49 (2015).
- 49) Davis BJ, Chapple CR, Sellers DJ, Naylor AL, Sillar D, Campbell A, Chess-Williams R. α1L-Adrenoceptors mediate contraction of human erectile tissue. J. Pharmacol. Sci., 137, 366–371 (2018).
- 50) Ishikawa H, Miller DD, Patil PN. Comparison of post-junctional alpha-adrenoceptors in iris dilator muscle of humans, and albino and pigmented rabbits. Naunyn Schmiedebergs Arch. Pharmacol., 354, 765–772 (1996).
- 51) Suzuki Y, Kanada A, Okaya Y, Aisaka K. Effect of JTH-601, a novel alpha(1)-adrenoceptor antagonist, on prostate function in dogs. Eur. J. Pharmacol., 394, 123–130 (2000).
- 52) Suzuki Y, Moriyama N, Okaya Y, Nishimatsu H, Kawabe K, Aisaka K. Age-related change of the role of alpha1L-adrenoceptor in canine urethral smooth muscle. Gen. Pharmacol., 33, 347–354 (1999).
- 53) Argyle SA, McGrath JC. An alpha(1A)/alpha(1L)-adrenoceptor mediates contraction of canine subcutaneous resistance arteries. J. Pharmacol. Exp. Ther., 295, 627–633 (2000).
- 54) Bagot K, Chess-Williams R. Alpha(1A/L)-adrenoceptors mediate contraction of the circular smooth muscle of the pig urethra. Auton. Autacoid Pharmacol., 26, 345–353 (2006).
- 55) Deplanne V, Galzin AM. Functional characterization of alpha-1-adrenoceptor subtypes in the prostatic urethra and trigone of male rabbit. J. Pharmacol. Exp. Ther., 278, 527–534 (1996).
- 56) Van der Graaf PH, Deplanne V, Duquenne C, Angel I. Analysis of alpha1-adrenoceptors in rabbit lower urinary tract and mesenteric artery. Eur. J. Pharmacol., 327, 25–32 (1997).
- 57) Hiraoka Y, Ohmura T, Sakamoto S, Hayashi H, Muramatsu I. Identification of alpha 1-adrenoceptor subtypes in the rabbit prostate. J. Auton. Pharmacol., 15, 271–278 (1995).
- 58) Hiraizumi-Hiraoka Y, Tanaka T, Yamamoto H, Suzuki F, Muramatsu I. Identification of alpha-1L adrenoceptor in rabbit ear artery. J. Pharmacol. Exp. Ther., 310, 995–1002 (2004).
- 59) Pennefather JN, Lau WA, Chin C, Story ME, Ventura S. Alpha(1L)-adrenoceptors mediate noradrenaline-induced contractions of the guinea-pig prostate stroma. Eur. J. Pharmacol., 384, 25–30 (1999).
- 60) Yamamoto Y, Koike K. Alpha1-adrenoceptors in the guinea pig thoracic aorta. J. Smooth Muscle Res., 35, 181–192 (1999).
- 61) Hirai T, Tsuru H, Tanimitsu N, Yajin K, Sasa M. Effect of JTH-601, a putative alpha(1L)-adrenoceptor antagonist, on guinea pig nasal mucosa vasculature. Eur. J. Pharmacol., 416, 141–144 (2001).
- 62) Stam WB, Van der Graaf PH, Saxena PR. Analysis of alpha 1L-adrenoceptor pharmacology in rat small mesenteric artery. Br. J. Pharmacol., 127, 661–670 (1999).
- 63) Fujimoto S. Alpha 1-adrenoceptor subtypes mediating contraction of the femoral artery in spontaneously hypertensive rats. Can. J. Physiol. Pharmacol., 72, 862–869 (1994).
- 64) Muramatsu I, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman AS, Tanaka T, Rodrigo MC, Myagmar BE, Simpson PC. Identification of alpha 1L-adrenoceptor in mice and its abolition by alpha 1A-adrenoceptor gene knockout. Br. J. Pharmacol., 155, 1224–1234 (2008).
- 65) Gray KT, Ventura S. Alpha1L-adrenoceptors mediate contractions of the isolated mouse prostate. Eur. J. Pharmacol., 540, 155–161 (2006).
- 66) Burt RP, Chapple CR, Marshall I. Alpha1A-adrenoceptor mediated contraction of rat prostatic vas deferens and the involvement of ryanodine stores and Ca2+ influx stimulated by diacylglycerol and PKC. Br. J. Pharmacol., 123, 317–325 (1998).
- 67) Ishida H, Saito SY, Dohi N, Ishikawa T. Mechanism of membrane depolarization involved in α1A-adrenoceptor-mediated contraction in rat tail and iliac arteries. Biol. Pharm. Bull., 42, 1741–1745 (2019).
- 68) Burt RP, Chapple CR, Marshall I. The role of capacitative Ca2+ influx in the alpha 1B-adrenoceptor-mediated contraction to phenylephrine of the rat spleen. Br. J. Pharmacol., 116, 2327–2333 (1995).
- 69) Lyles GA, Birrell C, Banchelli G, Pirisino R. Amplification of alpha 1D-adrenoceptor mediated contractions in rat aortic rings partially depolarised with KCl. Pharmacol. Res., 37, 437–454 (1998).
- 70) Patane MA, Scott AL, Broten TP, Chang RS, Ransom RW, DiSalvo J, Forray C, Bock MG. 4-Amino-2-[4-[1-(benzyloxycarbonyl)-2(S)- [[(1,1-dimethylethyl)amino]carbonyl]-piperazinyl]-6, 7-dimethoxyquinazoline (L-765,314): a potent and selective alpha1b adrenergic receptor antagonist. J. Med. Chem., 41, 1205–1208 (1998).
- 71) Doxey JC, Lane AC, Roach AG, Virdee NK. Comparison of the alpha-adrenoceptor antagonist profiles of idazoxan (RX 781094), yohimbine, rauwolscine and corynanthine. Naunyn Schmiedebergs Arch. Pharmacol., 325, 136–144 (1984).
- 72) Zheng W, Rampe D, Triggle DJ. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol. Pharmacol., 40, 734–741 (1991).
- 73) Tanaka Y, Imai T, Igarashi T, Takayanagi K, Otsuka K, Yamaki F, Tanaka H, Shigenobu K. Comparison of the Ca2+ entry channels responsible for mechanical responses of guinea-pig aorta to noradrenaline and thapsigargin using SK&F 96365 and LOE 908. Naunyn Schmiedebergs Arch. Pharmacol., 362, 160–168 (2000).
- 74) Danielsen PL, Rea SM, Wood FM, Fear MW, Viola HM, Hool LC, Gankande TU, Alghamdi M, Stevenson AW, Manzur M, Wallace HJ. Verapamil is less effective than triamcinolone for prevention of keloid scar recurrence after excision in a randomized controlled trial. Acta Derm. Venereol., 96, 774–778 (2016).
- 75) Lee K, Morita H, Iwamuro Y, Zhang XF, Okamoto Y, Nakagawa T, Hasegawa H, Furutani H, Miwa S, Masaki T. Pharmacological characterization of receptor-mediated Ca2+ entry in endothelin-1-induced catecholamine release from cultured bovine adrenal chromaffin cells. Naunyn Schmiedebergs Arch. Pharmacol., 360, 616–622 (1999).
- 76) Ou G, Fujisawa M, Yashiro A, Xu K, Yoshioka K, Obara K, Tanaka Y. Prostanoid TP receptor stimulation enhances contractile activities in guinea pig urinary bladder smooth muscle through activation of Ca2+ entry channels: Potential targets in the treatment of urinary bladder contractile dysfunction. Life Sci., 287, 120130 (2021).
- 77) Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br. J. Pharmacol. Chemother., 14, 48–58 (1959).
- 78) Wang Y, Deng X, Hewavitharana T, Soboloff J, Gill DL. Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin. Exp. Pharmacol. Physiol., 35, 1127–1133 (2008).
- 79) Yang XR, Lin MJ, McIntosh LS, Sham JS. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol., 290, L1267–L1276 (2006).
- 80) Yamagishi R, Akiyama K, Nakamura S, Hora M, Masuda N, Matsuzawa A, Murata S, Ujiie A, Kurashina Y, Iizuka K, Kitazawa M. Effect of KMD-3213, an alpha 1a-adrenoceptor-selective antagonist, on the contractions of rabbit prostate and rabbit and rat aorta. Eur. J. Pharmacol., 315, 73–79 (1996).
- 81) Murata S, Taniguchi T, Muramatsu I. Pharmacological analysis of the novel, selective alpha1-adrenoceptor antagonist, KMD-3213, and its suitability as a tritiated radioligand. Br. J. Pharmacol., 127, 19–26 (1999).
- 82) Tatemichi S, Kobayashi K, Maezawa A, Kobayashi M, Yamazaki Y, Shibata N. Alpha1-adrenoceptor subtype selectivity and organ specificity of silodosin (KMD-3213). Yakugaku Zasshi, 126, 209–216 (2006).
- 83) Moriyama N, Akiyama K, Murata S, Taniguchi J, Ishida N, Yamazaki S, Kawabe K. KMD-3213, a novel alpha1A-adrenoceptor antagonist, potently inhibits the functional alpha1-adrenoceptor in human prostate. Eur. J. Pharmacol., 331, 39–42 (1997).
- 84) Su TH, Morishima S, Suzuki F, Yoshiki H, Anisuzzaman AS, Tanaka T, Cheng JT, Muramatsu I. Native profiles of alpha(1A)-adrenoceptor phenotypes in rabbit prostate. Br. J. Pharmacol., 155, 906–912 (2008).
- 85) Morishima S, Tanaka T, Yamamoto H, Suzuki F, Akino H, Yokoyama O, Muramatsu I. Identification of alpha-1L and alpha-1A adrenoceptors in human prostate by tissue segment binding. J. Urol., 177, 377–381 (2007).
- 86) Freedman RR, Sabharwal SC, Desai N. Sex differences in peripheral vascular adrenergic receptors. Circ. Res., 61, 581–585 (1987).
- 87) Kafka MS, Lake CR, Gullner HG, Tallman JF, Bartter FC, Fujita T. Adrenergic receptor function is different in male and female patients with essential hypertension. Clin. Exp. Hypertens., 1, 613–627 (1979).
- 88) Yablonsky F, Riffaud JP, Lacolle JY, Dausse JP. Alpha 1- and alpha 2-adrenoceptors in the smooth muscle of male and female rabbit urethra. Eur. J. Pharmacol., 121, 1–8 (1986).
- 89) Takeda M, Hatano A, Komeyama T, Koizumi T, Mizusawa T, Kanai T, Tomita Y, Maruyama K, Nagatomo T. Alpha-1 adrenoceptor subtypes (high, low) in human benign prostatic hypertrophy tissue according to the affinities for prazosin. Prostate, 31, 216–222 (1997).
- 90) Nishimura A, Kitano K, Takasaki J, Taniguchi M, Mizuno N, Tago K, Hakoshima T, Itoh H. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc. Natl. Acad. Sci. U.S.A., 107, 13666–13671 (2010).
- 91) White CW, Short JL, Ventura S. Rho kinase activation mediates adrenergic and cholinergic smooth muscle contractile responses in the mouse prostate gland. Eur. J. Pharmacol., 721, 313–321 (2013).
- 92) Krautwurst D, Degtiar VE, Schultz G, Hescheler J. The isoquinoline derivative LOE 908 selectively blocks vasopressin-activated nonselective cation currents in A7r5 aortic smooth muscle cells. Naunyn Schmiedebergs Arch. Pharmacol., 349, 301–307 (1994).
- 93) Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores KE, Rink TJ. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J., 271, 515–522 (1990).
- 94) Drumm BT, Rembetski BE, Cobine CA, Baker SA, Sergeant GP, Hollywood MA, Thornbury KD, Sanders KM. Ca2+ signalling in mouse urethral smooth muscle in situ: role of Ca2+ stores and Ca2+ influx mechanisms. J. Physiol., 596, 1433–1466 (2018).
- 95) Prakriya M, Lewis RS. Store-operated calcium channels. Physiol. Rev., 95, 1383–1436 (2015).
- 96) Samanta A, Hughes TET, Moiseenkova-Bell VY. Transient receptor potential (TRP) channels. Subcell. Biochem., 87, 141–165 (2018).
- 97) Walker RL, Hume JR, Horowitz B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am. J. Physiol. Cell Physiol., 280, C1184–C1192 (2001).