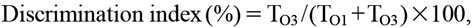MATERIALS AND METHODS
CellsHuman SH–SY5Y neuroblastoma cells (ATCC, Manassas, VA, U.S.A.) were seeded at density of 1.0 × 104 cells/cm2 on 10 cm plates and maintained in Eagle’s minimum essential medium (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan)/Ham’s F–12 (FUJIFILM Wako Pure Chemical Corporation) (1 : 1) containing penicillin–streptomycin solution (1×, FUJIFILM Wako Pure Chemical Corporation), non-essential amino acids solution (NEAA, 1×, FUJIFILM Wako Pure Chemical Corporation), L-glutamine solution (1×, FUJIFILM Wako Pure Chemical Corporation), and 15% fetal bovine serum (FBS, Sigma-Aldrich, St. Louis, MO, U.S.A.) in an incubator set at 37 °C, 5% CO2, and 95% air.
The human induced pluripotent stem cells (hiPSCs, line 1231A3) used in this study were provided by the RIKEN Bioresource Research Center (Tsukuba, Japan).33) The hiPSCs were maintained as previously described.34) Briefly, a 6-well-plate was coated with iMatrix-511 silk (Nippi, Tokyo, Japan), feeder-free hiPSCs were seeded at a density of 1.5 × 104 cells/well on the plate, and maintained in essential 8 (E8) medium (Thermo Fisher Scientific, Waltham, MA, U.S.A.) containing 10 µM Y–27632 (Selleck Chemicals, Houston, TX, U.S.A.) for the first day. Y–27632 was removed from the culture medium from day 2 onwards. The experimental protocols including hiPSCs were approved by the Ethical Review Committee for Medical and Health Research Involving Human Subjects at Kyoto Pharmaceutical University (Approval No. 20-18-26).
Generation of Cholinergic Neurons from hiPSCsThe hiPSCs were seeded at a density of 5.0 × 106 cells/well on iMatrix silk-coated 6-well-plate in E8 medium containing 10 µM Y–27632 and penicillin–streptomycin solution (1×) and cultured for 2 d. Differentiation toward cholinergic neurons began on the next day (day 0) using essential 6 (E6) medium containing a penicillin–streptomycin solution (1×), NEAA (1×), GlutaMAX (1×, Thermo Fisher Scientific), and a 2-mercaptoethanol solution (100 µM, FUJIFILM Wako Pure Chemical Corporation) as a basal medium. From day 0, cells were cultured with the E6 basal medium containing 10 µM Y–27632, 200 nM LDN193189 (Selleck Chemicals), 500 nM A83–01 (FUJIFILM Wako Pure Chemical Corporation), and 2 µM XAV939 (Selleck Chemicals). Purmorphamine (1 µM) was further added from day 1, and Y–27632 was removed from day 3. From days 5 to 11, the E6 basal medium was gradually replaced with neurobasal medium (Thermo Fisher Scientific) supplemented with B27 supplement vitamin A minus (1×, Thermo Fisher Scientific), GlutaMAX (1×, Thermo Fisher Scientific) including penicillin–streptomycin solution (1×), 200 nM LDN193189, 500 nM A83–01, 1 µM purmorphamine, and 2 µM XAV939. A83–01 was removed from the basal medium on day 6. From day 11, the cells were further maintained in culture medium without purmorphamine. On day 16, cells were detached with TrypLE (1×, Thermo Fisher Scientific), re-suspended and replated at a density of 1.0 × 106 cells/cm2 on culture plates coated with poly-L-ornithine hydrobromide (Sigma-Aldrich), iMatrix silk, and fibronectin (1 µg/mL; FUJIFILM Wako Pure Chemical Corporation) in neurobasal medium supplemented with B27 supplement vitamin A minus (1×), GlutaMax (1×), penicillin–streptomycin solution (1×), 5 µM SU5402 (Selleck Chemicals), 1 µM PD0325901 (Selleck Chemicals), 200 µM L-ascorbic acid, (Sigma-Aldrich), 10 µM DAPT (Selleck Chemicals), 20 ng/mL brain–derived neurotrophic factor (Peprotech, Rocky Hill, NJ, U.S.A.), and 10 µM Y–27632. Y–27632 was added for the first 24 h after replating. The medium was changed daily until day 15 and then every 3–4 d from day 16 onwards.
AnimalsWild-type C57BL/6 mice (10-week-old) were purchased from Oriental Bio Service (Kyoto, Japan). APdE9 transgenic mice, a mouse model of amyloid pathology, harboring transgenes for mutant Aβ precursor protein (AβPPswe: KM594/5NL) and presenilin 1 (dE9: deletion of exon 9), were originally purchased from the Jackson Laboratory (Bar Harbor, ME, U.S.A.). The transgenic mice were bred by mating with wild-type C57BL/6 mice. All mice were maintained at 25 °C under a 12-h light/dark cycle with free access to food and water at the Bioscience Research Center at Kyoto Pharmaceutical University. All animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Committee for Animal Research at Kyoto Pharmaceutical University (Approval No. DIPS-19-001).
Aβ Preparation and AggregationThe isoAβ (Peptide Institute Inc., Osaka, Japan) was dissolved in 0.1% TFA (FUJIFILM Wako Pure Chemical Corporation) and centrifuged at 20000 × g, 4 °C for 1 h. The protein concentration in the collected supernatant was determined by measuring absorption spectrum at 280 nm with a micro volume spectrophotometer (NanoPhotometer NP80, Implen, München, Germany). This diluted isoAβ stock (150 µM) was stored at –80 °C until further use. For Aβ aggregation, isoAβ was diluted with the culture medium for SH–SY5Y cells (containing 1% FBS) or phosphate-buffered saline (PBS) to a concentration of 6 µM and incubated at 37 °C.
Plant and Natural Product MaterialThe methanolic (MeOH) extract of B. monniera was prepared in a previous study.35) As described, dried whole plants of B. monniera cultivated in India were purchased from NTH India Pvt. Ltd. (2013) and identified by author H.M. A plant specimen is on file in Department of Pharmacognosy, Kyoto Pharmaceutical University [BM–NTH–1].
Isolation of an Aβ Binding Compound, Plantainoside B, from B. MonnieraBeads (Cytometric Bead Array Functional Bead A9, BD Biosciences, San Jose, CA, U.S.A.) used for isolation of Aβ binding compounds from the B. monniera extract were prepared as previously reported.36) Briefly, bead activation by labeling with an SH group was performed with 1 M dithiothreitol (DTT). In contrast, isoAβ (6 µM) was preliminarily incubated in PBS for 1 or 24 h and filtered (10 kDa, Amicon Ultra centrifugal filters, Merck Millipore, Darmstadt, Germany) to condense the aggregated Aβ. The free NH2 group in the aggregated Aβ (100 µL) was bounded to N-2[(4–maleimidomethyl)cyclohexylcarbonyloxy]sulfosuccinimide (sulfo–SMCC) (2 µL, 2 mg/mL in distilled water) at room temperature for 1 h. The SH group-labeled beads were resuspended in Coupling Buffer in Functional Bead Conjugation Buffer Set (20 µL, BD Biosciences, CA, U.S.A.) were mixed with sulfo-SMCC-conjugated Aβ aggregates (100 µL) and shaken for 1 h to allow the beads to bind to the Aβ aggregates. N-Ethylmaleimide was added to mask the remaining SH groups on the beads and shaken for another 15 min at room temperature. The beads conjugated with Aβ aggregates were then washed with Storage Buffer in Functional Bead Conjugation Buffer Set (BD Biosciences) and centrifuged at 900 × g for three min to remove the unbound Aβ. All reactions were performed in the dark.
The MeOH extract (100.0 g) was partitioned into an ethyl acetate (EtOAc)–H2O (1 : 1, v/v) mixture to obtain an EtOAc–soluble fraction (15.8 g, 15.8%) and H2O–soluble fraction (84.2 g, 84.2%). The EtOAc–soluble fraction (10 mg) was dissolved in 1 mL distilled water containing 2% DMSO and centrifuged at 10000 × g for 10 min. The resulting EtOAc–soluble fraction was collected and filtered (Cosmonice filter, 0.45 µm, Nacalai Tesque, Kyoto, Jajpan) to remove the insoluble components. The EtOAc–soluble fraction of B. monniera extract and the beads conjugated with Aβ aggregates were mixed and incubated for 1 h in the dark at room temperature; the beads were collected after centrifugation at 900 × g for three min. To separate the bound compounds, ethanol was added to the beads and placed in boiling water for five min. The identification of the compounds binding to Aβ aggregates was performed by LC/MS using a tandem quadrupole mass spectrometer (LCMS–8040, Shimadzu Corporation, Kyoto, Japan) equipped with Cosmosil MS–II column (5 µm particle size, 2.0 mm i.d. × 150 mm, Nacalai Tesque) and LabSolutions LCMS Ver.5.6 software (Shimadzu Corporation). For LC, water (pH adjusted to 3.0 using formic acid) and acetonitrile (MeCN) were utilized as mobile phases; the gradient started at 10% and increased to 100% after 1 h. MS conditions were optimized as follows: nebulizer gas flow, 3 L/min; drying gas flow, 15 L/min; desolvation line temperature, 250 °C; heat block temperature, 400 °C.
The above analysis identified plantainoside B as an Aβ binding compound in the EtOAc–soluble fraction of B. monniera. For isolation, the EtOAc–soluble fraction (14.0 g) was subjected to normal phase silica gel column chromatography [840 g, CHCl3:MeOH (30 : 1 → 15 : 1 → 7 : 1 → 5 : 1, v/v) → MeOH] resulting in ten fractions [Fr. E1 (2.2 g), Fr. E2 (1.6 g), Fr. E3 (0.3 g), Fr. E4 (0.4 g), Fr. E5 (0.9 g), Fr. E6 (1.5 g), Fr. E7 (4.8 g), Fr. E8 (0.7 g), Fr. E9 (0.8 g), Fr. E10 (0.9 g)]. Fr. E7 (1.1 g) was purified by HPLC [MeCN:H2O:acetic acid (CH3COOH) = 14 : 86 : 0.3, Cosmosil MS–II column (5 µm particle size, 10 mm i.d. × 250 mm, Nacalai Tesque)] to produce plantainoside B (45.9 mg, 0.228%). Plantainoside B was identified by comparing the measured physical data (MS and 1H- and 13C-NMR spectra) and reference data.37) NMR was performed using an NMR spectrometer [JEOL JNM–ECA600 (600 MHz), JEOL Ltd., Tokyo, Japan]. All chemicals used in above processes were purchased from Nacalai Tesque.
Blue Native–Polyacrylamide Gel Electrophoresis (BN–PAGE)The isoAβ aggregation was analyzed by BN–PAGE. Aβ aggregates produced after the incubation of isoAβ (6 µM) in the presence or absence of plantainoside B (100 µM) in culture medium or PBS at 37 °C for 1–72 h were mixed with a NativePAGE Sample Buffer (Thermo Fisher Scientific). The samples were then subjected to BN–PAGE (4–16% Bis–Tris mini protein gels; Thermo Fisher Scientific). After electrophoresis, immunoblotting was performed by transferring the proteins to a polyvinylidene difluoride (PVDF) membrane (Millipore). The PVDF membrane was incubated with Tris-buffered saline containing 0.05% Tween-20 (TBST) and Bullet Blocking One for Western blotting (Nacalai Tesque) to block nonspecific binding. The PVDF membrane was incubated with a primary antibody against Aβ (clone 6E10, 1 : 2000, Biolegend, San Diego, CA, U.S.A.) dissolved in Bullet Immunoreaction Buffer (Nacalai Tesque), followed by a horseradish peroxidase (HRP)-linked secondary antibody against mouse immunoglobulin G (IgG) (1 : 2000, Thermo Fisher Scientific) dissolved in Bullet Immunoreaction Buffer (Nacalai Tesque). HRP activity on the PVDF membrane was visualized with Chemi–Lumi One Super (Nacalai Tesque) and detected by a lumino–image analyzer (ImageQuant LAS500 system, GE Healthcare Life Science, Pittsburgh, PA, U.S.A.).
Aβ Cytotoxicity AssaySH–SY5Y cells were seeded at a density of 3.0 × 103 cells/well on a 96–well plate and cultured for 7 d, whereas hiPSC-derived cholinergic neuron progenitors at day 16 were seeded at a density of 1.5 × 105 cells/well on an iMatrix silk-coated 96–well plate and further cultured for 8–12 d. The culture medium of SH–SY5Y cells was replaced by medium containing 1% FBS before isoAβ treatment. Cells were treated with isoAβ (1–6 µM) in the presence or absence of plantainoside B (3–30 µM) for 48 h and Aβ cytotoxicity was analyzed through cell viability with the WST–8 assay and cytotoxicity with lactate dehydrogenase (LDH) assay. Both assays were performed using Cell Count Reagent SF (Nacalai Tesque) and LDH cytotoxicity assay kit (Nacalai Tesque) according to manufacturer’s instruction, respectively. In the LDH assay, the value obtained for the sample with cells lysed using the provided lysate reagent (Triton X-100) was set to 100% as the high control.
Immunocytochemical AnalysisCells were fixed in PBS containing 4% paraformaldehyde (PFA) for 30 min at 4 °C, then washed three times with PBS. The primary antibodies used in this study, Aβ (mouse, clone 6E10 1 : 500, Biolegend), NKX2.1 (rabbit, 1 : 500, Abcam, Cambrige, U.K.), FOXG1 (rabbit, 1 : 500, Abcam), ISLET–1 (rabbit, 1 : 500, Abcam), ChAT (goat, 1 : 250, Millipore), MAP2 (chicken, 1 : 1000, Abcam; mouse, 1 : 500, Sigma-Aldrich), and LHX8 (rabbit, 1 : 200, Thermo Fisher Scientific), were diluted in PBS containing 0.1% Triton X (PBST) and 5% donkey serum (Jackson ImmunoResearch) and incubated with fixed cells at 4 °C overnight. After washing with PBST, secondary antibodies (1 : 500 each): Alexa Fluor 488-labeled anti-rabbit IgG antibody (Thermo Fisher Scientific), Alexa Fluor 555-labeled anti-rabbit IgG antibody (Thermo Fisher Scientific), Alexa Fluor 647-labeled anti-rabbit IgG antibody (Abcam), Alexa Fluor 488-labeled anti-mouse IgG antibody (Thermo Fisher Scientific), Alexa Fluor 555-labeled anti-mouse IgG antibody (Thermo Fisher Scientific), Alexa Fluor 647-labeled anti-mouse IgG antibody (Thermo Fisher Scientific), Alexa Fluor 555-labeled anti-goat IgG antibody (Thermo Fisher Scientific), and Alexa Fluor 488-labeled anti-chicken IgY antibody (Abcam) in PBST were incubated with the cells for 2 h at room temperature in the dark. Hoechst 33342 (1 : 4000, Thermo Fisher Scientific) and rhodamine–phalloidin (1 : 3000, Thermo Fisher Scientific) for staining the nuclei and actin cytoskeleton, respectively, were added in the secondary antibody solution. Fluorescence was detected with a confocal laser scanning microscope (LSM800, Zeiss, Oberkochen, Germany).
Calcium ImagingOn day 16, hiPSC-derived cholinergic neuron progenitors were seeded at a density of 3.0 × 105 cells/well in a 4-well plate (Matsunami, Osaka, Japan) coated with iMatrix silk and cultured for another 8–10 d. Cells were treated with fluo–4 AM (10 µM, Thermo Fisher Scientific), a fluorescent calcium indicator dye, incubated at 37 °C for 30 min; then, the culture was replaced with fresh culture medium without fluo-4 AM. The isoAβ (3 µM) with or without plantainoside B (30 µM) was added to the cells. Following incubation for 2 h, the cells were placed in an incubation chamber at 37 °C, 5% CO2, and 95% air for microscopic analysis, and time–lapse imaging was performed to detect fluo-4 fluorescence with a confocal laser scanning microscope (LSM800). Fluorescent images were acquired every five min for 3 h.
Measurement of Mitochondrial Membrane Potential (MMP) by JC–1 AnalysisMMP was analyzed using the JC–1 dye (JC–1 MitoMP Detection Kit, Dojindo, Kumamoto, Japan). JC–1, as a cationic fluorescent dye, exhibits potential–dependent accumulation in the mitochondria, forming aggregates (red fluorescent). Upon depolarization, JC–1 diffuses across the mitochondria in monomeric state (green fluorescent).38) Thus, a higher ratio of red/green fluorescence indicates that the mitochondria are energetically active, whereas a lower ratio indicates MMP loss.
The hiPSC-derived cholinergic neurons were seeded as in the cytotoxicity assay and treated with isoAβ (3 µM) with or without plantainoside B (3–30 µM) for 48 h. After washing with new culture medium, JC–1 dye (2 µM in final concentration) was added to the cells and incubated at 37 °C, 5% CO2, and 5% O2 for 30 min. After several washes, culture medium was replaced by the Imaging Buffer provided in the assay kit, and green and red fluorescence were measured with filter sets of excitation (475 nm)/emission (500–550 nm) and excitation (520 nm)/emission (580–640 nm), respectively using a fluorescence plate reader (GloMAX, Promega, Madison, U.S.A.). The red/green fluorescence ratio was calculated afterwards.
Thioflavin-T (ThT) Fluorescence AssayThe isoAβ (6 µM) was dissolved in PBS in the presence or absence of plantainoside B (100 µM) and incubated at 37 °C, 5% CO2, and 95% air for 1–24 h. After incubation, ThT (10 µM in final concentration) was added to the samples and the fluorescence spectra obtained under excitation at 485 nm were recorded using a fluorescence spectrophotometer (F–7000, Hitachi, Tokyo, Japan).
Atomic Force Microscopy (AFM)Aβ aggregation was analyzed by AFM. The isoAβ (6 µM) was dissolved in PBS with or without plantainoside B (100 µM) in Protein LoBind Tubes (Eppendorf, Hamburg, Germany) and incubated at 37 °C, 5% CO2, and 95% air for 24 h. After incubation, Aβ aggregates were diluted with distilled water (1 : 10). The Aβ solution (20 µL) was spotted onto freshly cleaved mica (Nilaco Corp., Tokyo, Japan) and incubated for 10 min at room temperature. After washing the mica with distilled water, images were obtained under ambient conditions at room temperature using Nanoscope IIIa Tapping Mode AFM (Veeco Instrument, Plainview, NY, U.S.A.) and a single–crystal microcantilever (OMCLAC160TS–R3, Olympus, Tokyo, Japan). At least three regions of each sample were analyzed.
Image AnalysisFor the measurement of the Aβ covered area on the cell surface, confocal laser scanning microscope (LSM800) Z-stack images were analyzed, and Aβ attached to the cells was measured using an image analysis software (Photoshop, version 22.2.0., Adobe, San Jose, CA, U.S.A.) using the fluorescence of actin and Aβ detected by rhodamine–phalloidin and anti-Aβ antibody as reference, respectively. The percentage of Aβ covered area on the cell surface was then calculated.
The number of cells expressing cholinergic markers, such as LHX8, ISLET-1, and ChAT, was counted using an image analysis software (Photoshop) for the confocal laser scanning microscopy images (LSM800). Nuclei stained with Hoechst 33342 were counted and considered as the number of total cells. The percentage of cells expressing each cholinergic marker was subsequently calculated using the values indicated above.
Radiosynthesis of 125I-Labeled Plantainoside BSodium [125I]iodide ([125I]NaI, carrier free) solution was purchased from PerkinElmer, Inc. (Waltham, MA, U.S.A.) LD-20AD (Shimadzu, Kyoto, Japan) was used for reversed-phase (RP)-HPLC, along with an SPD-20A (Shimadzu) UV detector (220, 254 nm) and RI analyzer Gabi Nova (Elysia-raytest, Liege, Belgium). Radiosynthesis of 125I-labeled plantainoside B was performed by electrophilic aromatic substitution reaction. Plantainoside B (500 µg) dissolved in MeOH (10 µL) was mixed with a solvent (MeOH:CH3COOH = 70 : 20, 90 µL) containing N-chlorosuccinimide (0.5 mg). [125I]NaI (15 MBq) was added to the mixture, and then vortexed at room temperature for 30 min. After removing the solvent using N2 gas, the mixture was purified through RP-HPLC using Cosmosil 5C18-AR-II column (4.6ID × 150 mm, Nacalai Tesque) with H2O:MeCN (gradient from 10% to 50% MeCN over 30 min) as the mobile phase at a flow rate of 1.0 mL/min, to obtain 125I-labeled plantainoside B (3.2–5.9 MBq). MeCN was removed from the sample using N2 gas and the radio activity was measured using a dose calibrator (IGC-8, Hitachi Ltd., Tokyo, Japan).
Autoradiography and Immunohistochemical AnalysisBrain sections from male 9-month-old APdE9 mice and littermate wild-type mice were used for autoradiography. Mice were euthanized through cervical dislocation and the brains were immediately removed and fixed with 4% PFA at 4 °C for 3 d, followed by dehydration with 30% sucrose for 3 d. Brains were then sectioned (20 µm thickness) using a cryostat. Brain sections were incubated with 125I–labeled plantainoside B solution (adjusted to 350 kBq/mL using PBS) on a shaker at room temperature for 2 h, washed three times with PBS, and dried completely on a slide glass (Matsunami, Osaka, Japan). Alternatively, Aβ aggregates resulting from the incubation of isoAβ (6 µM) in PBS at 37 °C for 1 and 6 h were mixed with 125I–labeled plantainoside B solution (adjusted to 20 MBq/mL with PBS); the samples were then run on blue native–polyacrylamide gels (4–16% Bis–Tris mini protein gels; Thermo Fisher Scientific). The brain sections and BN–PAGE gels were exposed to a phosphor–imaging plate (FUJIFILM Plate BAS–TR2025, FUJIFILM, Tokyo, Japan) overnight. The imaging plate was scanned using an image analyzer (Typhoon 9410, GE Healthcare, Milwaukee, WI, U.S.A.) at a resolution of 25 μm/pixel.
The brain sections were also incubated with anti-Aβ antibody (clone 6E10, 1 : 2000) and Hoechst 33342 (1 : 4000) overnight at room temperature, followed by incubation with secondary antibody, Alexa Fluor 555-labeled anti-mouse IgG antibody (1 : 500), for 2 h. Fluorescence was detected using a confocal laser scanning microscope (LSM800).
Intrahippocampal Injection of isoAβTen-week-old wild-type male C57BL/6 mice were anesthetized by intraperitoneal injection with a mixture of medetomidine (0.3 mg/kg, Domitor, ZENOAQ, Koriyama, Japan), midazolam (4 mg/kg, Dormicum, Maruishi Pharmaceutical Co., Osaka, Japan), and butorphanol (5 mg/kg, Vetorphale, Meiji Seika Pharma, Tokyo, Japan). Mice were placed on a stereotaxic instrument (51730D; Stoelting, Wood Dale, IL, U.S.A.) and injected with PBS, isoAβ (20 µM), or isoAβ (20 µM) plus plantainoside B (30 µM) at a total volume of 3 µL PBS solution into the bilateral hippocampi (coordinates from bregma: ± 1.5 mm lateral, 2.0 mm posterior, and 2.0 mm ventral) using a 10 µL Hamilton syringe driven by an automated syringe pump (set at 1 µL/min injection rate, Legato130, KD Scientific, Holliston, MA, U.S.A.). After administration, the syringe was slowly removed from the brain and the skin was sutured. Mice received atipamezole (0.3 mg/kg, Antisedan, Orion Co., Espoo, Finland) and returned to the warm box to recover from anesthesia.
Cognitive Assessment with Novel Object Recognition Test (NORT)Cognitive performance was evaluated with NORT. Mice were placed in the open–field box (length × width × height: 40 × 40 × 30 cm) under a light set at 20 lx and habituated for 10 min. Thirty minutes after habituation, two objects (O1 and O2) were presented to mice for 10 min (trial phase). After a 24 h interval for memory retention, the mice were placed in the same box again where the O2 object was replaced by a novel third object (O3); object preference was analyzed during the test phase for 10 min. Behavioral movements were recorded for 10 min during the test phase with a video camera connected to PC with a tracking system (EthoVision XT 11.5, Noldus Information Technology). An 8 × 8 cm square (target area) centered on each object was set up, and the time spent in the target area (T) was measured. The discrimination index, used as a measure of nonspatial working memory function, was calculated as follows:
where TO1 and TO3 are the time spent in the O1 or O3 target areas during the test phase, respectively.
Statistical AnalysesGraphPad Prism 5 (GraphPad Software, La Jolla, CA, U.S.A.) was used for all statistical analyses. Results are presented as means ± standard errors of the means (S.E.Ms.). Student’s t-test was used for two-group comparisons. For multiple comparisons, the one-way ANOVA followed by a Bonferroni’s post-hoc multiple comparison test was used for comparing among ≥3 groups. A p–value <0.05 was considered statistically significant.
RESULTS
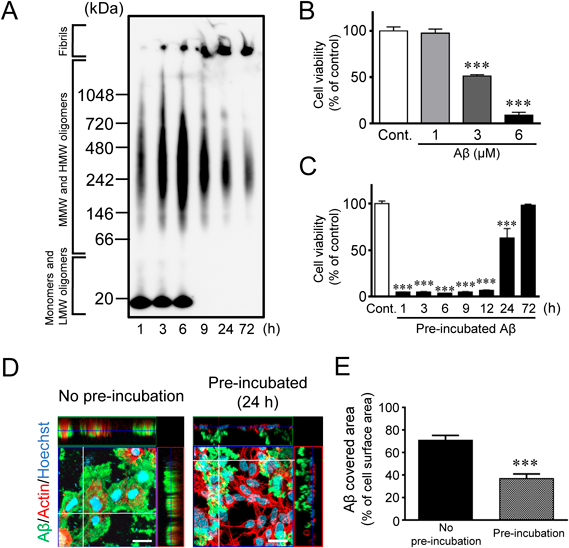
The isoAβ Aggregation and Cell Toxicity Induced by Aβ OligomersWe first attempted to construct an Aβ oligomer-dependent neurodegeneration model system in vitro. To this end, isoAβ, which reverts to natural Aβ1−42 and initiates self-aggregation under neutral pH conditions, was used for stable and uniform Aβ oligomer formation.24) In the present study, the time-dependent aggregation of isoAβ under neutral pH conditions at 37 °C for 1–72 h was analyzed by BN–PAGE (Fig. 1A). Aβs around 20 kDa (low molecular weight Aβ oligomers and isoAβ monomer) were stably detected up for to 6 h after incubation but not after 9 h. Similarly, Aβ aggregates from about 66 kDa to over 1048 kDa (middle to high molecular weight Aβ oligomers) increased after up to 6 h of incubation, gradually decreasing after 9 h. In contrast, the Aβ fibrils that did not enter the separation gel were clearly detected in the sample incubated for 3 h; their amount increased in a time–dependent manner over incubation.
Subsequently, we examined cell viability after isoAβ treatment with a WST–8 assay using SH–SY5Y cells, a neuroblastoma cell line, cultured under neutral pH conditions. Direct treatment with isoAβ for 48 h significantly decreased SH–SY5Y cell viability in a concentration–dependent manner (Fig. 1B). Next, to narrow down the type of Aβ aggregates responsible for decreased cell viability, aggregates formed after various pre-incubation times were added to SH–SY5Y cells (Fig. 1C). The viability of SH–SY5Y cells was significantly reduced by treatment with Aβ aggregates made by pre-incubation < 12 h whereas those Aβ aggregates resulting from > 24 h pre-incubation caused less or almost no change in cell viability (Fig. 1C). Confocal laser scanning microscopic analysis revealed that Aβ was tightly attached on the surface of SH–SY5Y cells when cells were directly treated with isoAβ without pre-incubation (Figs. 1D, E). In contrast, treatment with Aβ aggregates, produced by pre-incubation in culture medium under neutral pH for 48 h, showed much less binding to the cell surface (Figs. 1D, E). Thus, as shown by this comparison, the non-pre-incubated Aβ adhered strongly to the cell surface, whereas the pre-incubated Aβ adhered weakly to some cells, perhaps also to the bottom of the culture dish, and mostly remained suspended in the cell culture medium. Based on these results, Aβ oligomers (about 66 kDa to over 1048 kDa), but not isoAβ monomers, low molecular weight Aβ oligomers (around 20 kDa), or fibrils, may be involved in the reduced cell viability through tight attachment to the cell surface in this model.
Differentiation of Basal Forebrain Cholinergic Neurons from hiPSCs and isoAβ Induced NeurodegenerationTo develop a specific neurodegeneration model based on the cholinergic hypothesis in AD,2) we investigated the differentiation of cholinergic neurons targeting the basal forebrain from hiPSCs. Recently, several methods for differentiating cholinergic neurons from pluripotent stem cells have been reported.3–5) Similarly, we independently developed a feeder-free differentiation protocol for basal forebrain and cholinergic neurons using low molecular weight compounds (small molecules) and confirmed the differentiation process (Figs. 2A, B). Confocal laser scanning microscopy revealed the expression of FOXG1 (a telencephalic marker), NKX2.1 (a ventral telencephalic marker),39) and ISLET–1 (a cholinergic neuron maker)5) in iPSC-derived cells on day 16 after induction (Fig. 2A). On day 24, the differentiation toward cholinergic neurons further progressed, and expression of ChAT and LHX8 (cholinergic neuron markers)39) and MAP2 (a dendrite marker)34) was detected (Fig. 2B). Based on the immunoreactivity of cholinergic neuron markers such as LHX8, ISLET–1, and ChAT, detected on day 24 of differentiation, the percentage of cholinergic neurons to the total cells (Hoechst33342+ cells) was calculated (Fig. 2B). Although the rates of transcription factors and early markers of cholinergic neurons,40) such as LHX8 (25.3 ± 7.4%) and ISLET–1 (47.7 ± 5.1%), were relatively low, most cells were positive for ChAT (86.6 ± 3.1%), which is the synthetic enzyme for acetylcholine (Fig. 2B); these results suggest that the cells differentiated beyond the initial stages and are capable of functioning as cholinergic neurons. Thus, we successfully differentiated basal forebrain cholinergic neurons from hiPSCs with a simplified protocol.
We next examined isoAβ induced neurodegeneration using hiPSC-derived cholinergic neurons on day 24. Cell viability and cytotoxicity were analyzed by the WST–8 (Fig. 2C) and LDH assays (Fig. 2D), respectively. Similar to SH–SY5Y cells, the isoAβ treatment induced neurodegeneration of hiPSC-derived cholinergic neurons in a concentration-dependent manner. Confocal laser scanning microscopy revealed that Aβ was tightly bound to the surface of both cell bodies (Fig. 2E) and neurites (Fig. 2F) of hiPSC-derived cholinergic neurons generated after 24 d of differentiation. Thus, using iPSC-derived cholinergic neurons and isoAβ, we constructed a neurodegeneration model based on the cholinergic and Aβ oligomer hypothesis.
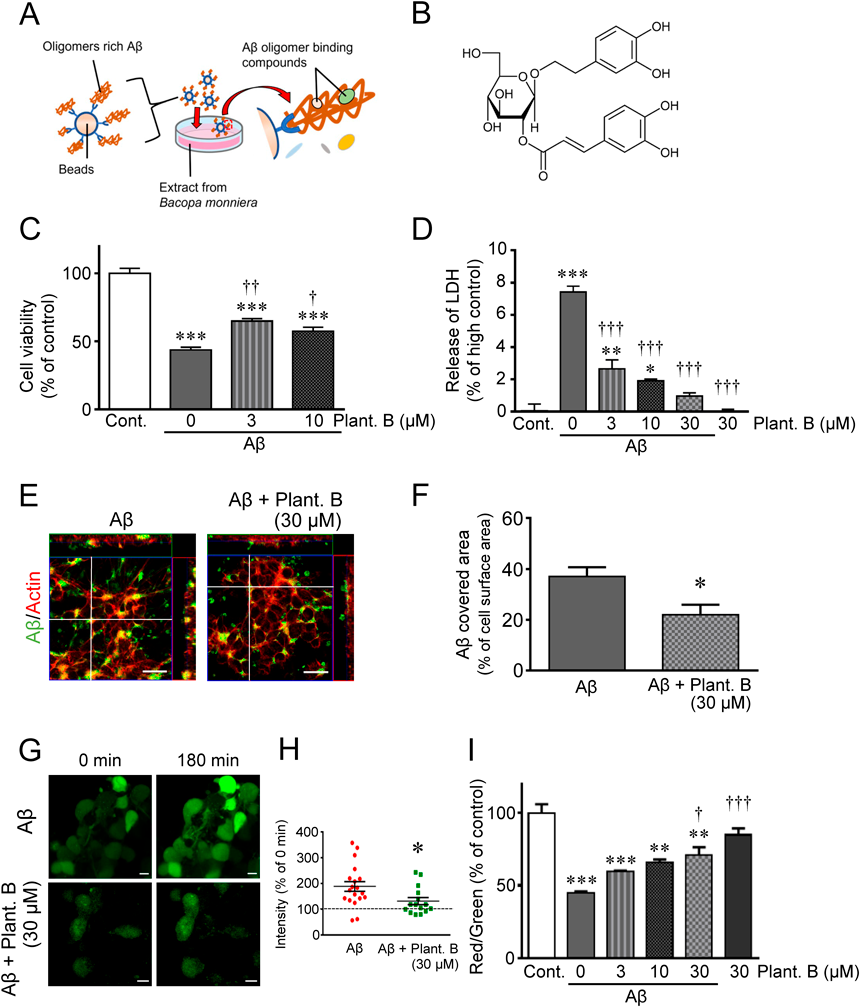
Neuroprotection by Plantainoside B, an Aβ Oligomer–Binding Small Molecule Isolated from B. Monniera ExtractWe next attempted to identify Aβ oligomer binding small molecules with potential neuroprotective activity (Fig. 3A). Beads conjugated with Aβ aggregates produced after 1 or 24 h of isoAβ incubation were immersed in the EtOAc fraction of B. monniera extract. LC/MS revealed that several compounds derived from B. monniera extract were bound to Aβ aggregates. Of these, small molecules (molecular weight < 500) that bound more to the Aβ aggregates produced by 1 h incubation than that of 24 h were chosen, and plantainoside B was identified (molecular weight; 478.45) by NMR and MS analysis (Fig. 3B).
We subsequently analyzed the neuroprotective activity of plantainoside B in the neuronal cell death model described above using isoAβ and hiPSC-derived cholinergic neurons. Plantainoside B significantly attenuated the decreased cell viability (Fig. 3C) and increased the LDH release (Fig. 3D) induced by Aβ oligomers in a concentration-dependent manner.
To explore the neuroprotective mechanism of plantainoside B, we performed confocal laser scanning microscopy. Plantainoside B treatment reduced Aβ binding to hiPSC-derived cholinergic neurons (Figs. 3E, F). On the other hand, Aβ oligomers induce Ca2+ influx into the cytoplasm.41–44) We, therefore, examined Ca2+-imaging using a fluorescent dye (fluo–4) and found that plantainoside B suppressed the Aβ-induced Ca2+ influx in hiPSC-derived cholinergic neurons (Figs. 3G, H). Moreover, as MMP dysregulation by Aβ oligomers has been reported,45) we further evaluated MMP using the indicator dye, JC–1.38) We found that plantainoside B significantly attenuated Aβ-induced MMP dysregulation in hiPSC-derived cholinergic neurons (Fig. 3I). Taken together, these findings suggest that plantainoside B may attenuate neurodegeneration by blocking the binding of Aβ oligomers to the cell surface, thereby suppressing subsequent cytotoxic events such as dysregulation of the cytoplasmic Ca2+ level and MMP.
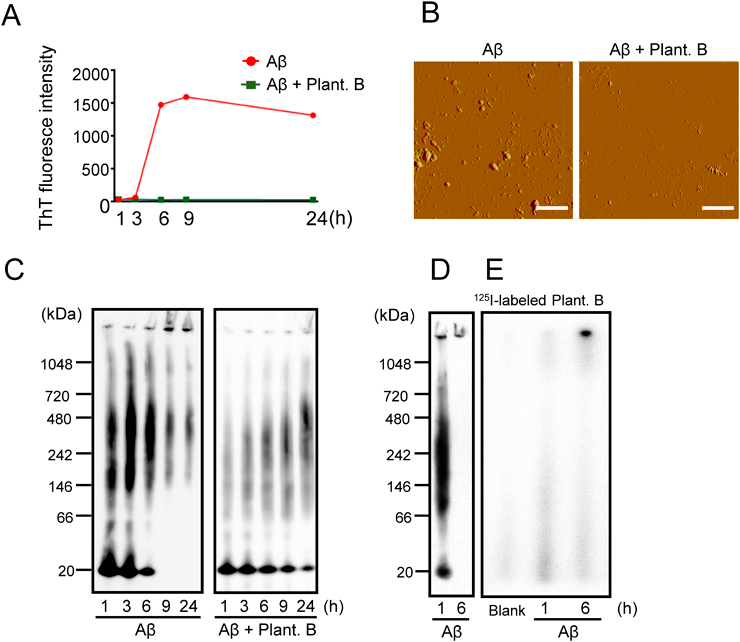
Inhibitory Effect of Plantainoside B on Aβ Aggregation and Binding Affinity to Aβ OligomersWe next examined the effects of plantainoside B on Aβ aggregations. A ThT assay revealed that isoAβ markedly self-aggregated after 6 h of incubation and that plantainoside B strongly suppressed aggregation (Fig. 4A). We further examined the inhibitory effect of plantainoside B on Aβ aggregation by AFM (Fig. 4B) and BN–PAGE (Fig. 4C); both confirmed the suppressive effect of plantainoside B on Aβ aggregation.
The binding affinity of plantainoside B to Aβ aggregates was analyzed using BN–PAGE (Figs. 4D, E). Aβ aggregation was first confirmed by BN–PAGE with anti-Aβ antibody (Fig. 4D). A smear band of Aβ aggregates (from around 20 kDa Aβs to high molecular fibrils) was detected 1 h after isoAβ incubation in PBS whereas a band of fibrils was detected 6 h after incubation (Fig. 4D). Subsequently, 125I–labeled plantainoside B was run on blue native–polyacrylamide gels with or without Aβ aggregates made by pre-incubation of isoAβ for 1 or 6 h in PBS. In autoradiography, despite background radioactivity of 125I–labeled plantainoside B on the blank (125I–labeled plantainoside B only), Aβ pre-incubated for 1 h at the position with Aβ aggregates (Aβs from around 20 to 242 kDa and fibrils over 1048 kDa) (Fig. 4E) produced a stronger signal. On the other hand, weaker and stronger signals were similarly detected with Aβ pre-incubated for 6 h with Aβ aggregates (around 20 to over 242 kDa) and fibrils, respectively (Fig. 4E). Because of the high sensitivity of radioactivity, 125I–labeled plantainoside B could be detected in Aβ aggregates that could not be detected with the anti-Aβ antibody in the sample with Aβ pre-incubated for 6 h. Accordingly, plantainoside B appears to have binding affinity to Aβ aggregates (especially, from around 20 to over 242 kDa) and fibrils.
Binding Affinity of Plantainoside B to Brain Sections from Mice with Aβ Pathology and Attenuation of Memory Impairment in IsoAβ Injected MiceWe further examined the binding affinity of 125I–labeled plantainoside B to brain sections from a wild-type mouse and a model mouse of Aβ pathology (APdE9). Confocal laser scanning microscopic analysis revealed Aβ accumulation in the brain tissue from model mice but not from wild-type mice (Fig. 5A). In the autoradiography, diffuse and much stronger 125I–labeled plantainoside B signals were detected in the brain sections of model mice compared with those of wild-type mice (Fig. 5B). In contrast, treatment with 125I− did not get clear radioisotope signals in the brain tissues from a wild-type mouse and a mouse model of amyloid pathology (Supplementary Fig. S1). Moreover, blocking with an excess amount of non-labeled plantainoside B compared with that of 125I-labeled plantainoside B reduced the radioactivity in the brain tissues from a mouse model of amyloid pathology (Supplementary Fig. S1). Thus, the high radioactivity in brain tissues from a mouse model of amyloid pathology obtained after incubation with 125I-labeled plantainoside B (Fig. 5B) was considered to be specific to the binding affinity of plantainoside B rather than 125I.
Finally, we examined the effects of plantainoside B on Aβ-induced memory deficits in wild–type mice. Mice were bilaterally injected with PBS or isoAβ with or without plantainoside B into the hippocampus and NORT was performed to investigate memory function (Fig. 5C). The intrahippocampal injection of isoAβ induced memory deficits as indicated by the discrimination index (Fig. 5D) and trajectories (Fig. 5E). In contrast, simultaneous injection with plantainoside B attenuated the memory deficits induced by the intrahippocampal injection with isoAβ (Figs. 5D, E). Thus, plantainoside B improved the Aβ-induced memory deficits in mice.
DISCUSSION
In familial AD (FAD) with the Arctic mutation (E693G) in AβPP, there is a high production of Aβ protofibrils.46) In FAD with the Osaka variant (E693Δ) of AβPP, the mutant Aβ (E22Δ) primarily produces Aβ oligomers rather than Aβ fibrils. Thus, the genetic analysis of FAD strongly supports that the neurotoxicity of Aβ oligomers is responsible for the neurological symptoms in AD.47,48) In the present study, we found that the neurotoxicity of isoAβ decreases as the amount of Aβ monomers and oligomers decreased and fibrils gradually formed (Figs. 1A, C). Furthermore, significant cytotoxicity was detected (Fig. 1C) when monomers and low molecular weight Aβ oligomers < 20 kDa were barely detectable after 9 h of incubation (Fig. 1A). However, these results do not rule out the neurotoxicity of monomers and low molecular weight Aβ oligomers, rather suggest that middle and high molecular weight Aβ oligomers (about 66 kDa to over 1048 kDa) such as ADDLs, AβO, and protofibrils are the primary neurotoxic forms in the present neurodegeneration model.
In the present study, we further detected tight binding of Aβ to the cell surface, significant increase of cytosolic Ca2+ levels, and MMP disruption in hiPSC-derived cholinergic neurons after treatment with Aβ oligomers, confirming that human basal forebrain cholinergic neurons are indeed impaired by Aβ oligomers. The increase of cytosolic Ca2+ levels and mitochondrial dysfunction caused by Aβ oligomers was previously demonstrated in primary cultured-human fetal cortical neurons and rat cortical neurons.17,49) Regarding the mechanisms of increased cytosolic Ca2+ levels by treatment with Aβ oligomers, Ca2+ channels produced by Aβ, called amyloid channels,50,51) and activation of N-methyl-D-aspartate receptors44,52) have been implicated. Moreover, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors are involved in the Aβ oligomer-induced Ca2+ influx into the cytoplasm.53) However, these studies were performed in immortalized cell lines and primary cultured rodent cortical and hippocampal neurons. Therefore, the Aβ oligomer-induced neurodegeneration model constructed in the present study is advantageous in that the above mechanism could be analyzed using human basal forebrain cholinergic neurons in the future.
Impairment of cholinergic neurons in the nucleus basalis of Meynert is an early pathology of AD.1,2) A study using a mouse model of Aβ pathology showed that the activation of the cholinergic circuit from the basal forebrain to the hippocampus can improve the cholinergic metabolism and attenuate memory impairment in mice.54) Therefore, early protection of cholinergic neurons in the basal forebrain is key to cure AD and elucidating the pathological mechanism underlying the cholinergic neurodegeneration induced by Aβ, especially Aβ oligomers, is expected to contribute to the development of DMTs in AD. However, the molecular mechanisms of Aβ oligomer–induced neurodegeneration in human cholinergic neurons are not fully understood due to the difficulty of handling unstable Aβ oligomers55) and the lack of appropriate models for human cholinergic neurons.56,57) In the present study, we attempted to overcome this problem by combining the isoAβ27,28) with hiPSC technologies. The isoAβ reproduces well Aβ oligomers and has been used to analyze the toxicity of Aβ oligomers.25) Furthermore, FAD studies using isoAβ revealed abnormal Aβ oligomerization with the A673V mutation of AβPP.24) With respect to the hiPSC technologies, differentiation protocols of cholinergic neurons from mouse and human pluripotent stem cells have been reported.3–5) In the present study, we also independently developed a method for the differentiation of hiPSCs into cholinergic neurons targeting the basal forebrain using small molecules under the feeder-free condition. While previous reports have confirmed the expression of ChAT, a cholinergic neuronal marker, at more than 40 d after differentiation, in our protocol, ChAT was detected at 24 d after differentiation. In addition to the time savings, replacing humoral factors with small molecules in differentiation process could have resulted in a cost-effective and more stable protocol. Thus, the elucidation of the cell death mechanism using isoAβ with hiPSC-derived basal forebrain cholinergic neurons can assist the development of diagnosis methods and DMTs based on the Aβ oligomer and cholinergic hypothesis.
Plantainoside B has been shown to reduce plasma triglycerides levels in mice.58) However, its effects on neurons have not been previously reported. In the present study, we found for the first time that plantainoside B has an affinity for a broad range of Aβ aggregates (Aβs from around 20 kDa to 242 kDa and fibrils over 1048 kDa) and neuroprotective activity. This binding affinity of plantainoside B to Aβ oligomers may critically contribute to the inhibition of Aβ binding to the cells and slow the rate of Aβ aggregation. The inhibition of Aβ oligomer binding to the cell surface may be a primary mechanism of neuroprotection as indicated by the attenuated increase of cytosolic Ca2+ level and MMP dysregulation. Previously, a B. monniera extract was shown to significantly reduce the total levels of Aβ1–42 in the brain of a mouse model of amyloid pathology.31) It is possible that the binding affinity of plantainoside B in B. monniera extract to Aβ aggregates and its aggregation–delaying action may decrease the levels of Aβ in the brain, but the extent of this contribution is unknown. Therefore, research on the administration of a single component of plantainoside B to mouse models of Aβ pathology is needed in the future.
While technological development toward in vitro diagnosis for AD by the detection of Aβ in blood and cerebral spinal fluid continues,59,60) a significance of bioimaging is that it can detect pathogenic factors in the body as well as modulate them in the host. This makes bioimaging a useful tool in theranostics. Theranostics aims to influence the pathogenesis mechanism by initiating early treatment along with diagnosis, delaying the onset of disease or reducing symptoms, thereby enhancing treatment effectiveness and improving the patient’s QOL; this concept has already been adopted in oncology.61,62) In a similar, for neurodegenerative diseases and neurological disorders including AD, where early diagnosis and treatment are required, introducing theranostics (neurotheranostics) as a method of intervention would be highly beneficial.63,64) Since the development of Pittsburgh Compound-B, a positron emission tomography tracer for amyloid imaging,65) research has been conducted to develop further non-invasive diagnostic tools for AD, such as probes for magnetic resonance imaging (MRI). Curcumin, a polyphenol derived from the natural herb turmeric, directly inhibits fibrillation of Aβ and destabilizes Aβ aggregates,66) and curcumin derivatives are being developed as Aβ oligomer–specific probes for MRI with neuroprotective properties.67,68) Furthermore, rutin, a natural flavonol glycoside in various plants, has been combined with Congo red and magnetic nanoparticles to develop MRI probes for amyloid plaques with antioxidant properties.69) In the present study, plantainoside B showed neuroprotective and inhibitory effects on memory deficits induced by Aβ. Furthermore, radioisotope-labeled plantainoside B detected Aβ aggregates including its oligomers on blue–native polyacrylamide gels and produced a diffuse radioactive signal in the brain sections of a mouse model of amyloid pathology. Of note, the diffuse nature of the signal detected in the brain sections may be attributed to the fact that plantainoside B detected Aβ oligomers distributed throughout the brain sections, rather than only the Aβ fibrils that constitute Aβ plaques. Taken together, these results suggest that plantainoside B may be a pioneer compound that provides an opportunity for the development of theranostics in AD research. Accordingly, this study may provide valuable information for the practical application of theranostics, which is a new concept in AD treatment.
In summary, we established a neurodegeneration model specialized to the Aβ oligomer and cholinergic hypothesis in AD using isoAβ and hiPSC-derived basal forebrain cholinergic neurons. Using this model, we found that plantainoside B, a compound in B. monniera, shows neuroprotective effects by having binding affinity to Aβ aggregates. Plantainoside B further showed a high binding affinity to brain tissue from a mouse model of amyloid pathology and attenuated Aβ induced memory deficits in mice. Thus, the binding capacity to Aβ aggregates and neuroprotective effects of plantainoside B indicate its potential as a novel neurotheranostics tool for AD that could accelerate the development of new diagnostic methods and DMTs.