Note
Molecular Mechanism of SLC6A8 Dysfunction with c.1699T > C (p.S567P) Mutation in Cerebral Creatine Deficiency Syndromes
2024 年 47 巻 1 号 p. 187-191
詳細
2024 年 47 巻 1 号 p. 187-191
Cerebral creatine deficiency syndromes (CCDS) are neurodevelopmental disorders caused by a decrease in creatine levels in the central nervous system (CNS) due to functional mutations in creatine synthetic enzymes or creatine transporter (CRT/SLC6A8). Although SLC6A8 mutations have been reported to be the most frequent cause of CCDS, sufficient treatment for patients with CCDS harboring SLC6A8 mutations has not yet been achieved. This study aimed to elucidate the molecular mechanism of SLC6A8 dysfunction caused by the c. 1699T > C missense mutation, which is thought to induce dysfunction through an unidentified mechanism. A study on SLC6A8-expressing oocytes showed that the c.1699T > C mutation decreased creatine uptake compared to that in wild-type (WT) oocytes. In addition, a kinetics study of creatine uptake revealed that the c.1699T > C mutation reduced the maximum uptake rate but not Michaelis–Menten constant. In contrast, the c.1699T > C mutation did not attenuate SLC6A8 protein levels or alter its cellular localization. Based on the SLC6A8 structure in the AlphaFold protein structure database, it is possible that the c.1699T > C mutation alters the interaction between the S567 and Y143 residues of SLC6A8, leading to decreased creatine transport function. These findings contribute to the understanding of the pathology of CCDS and to the development of strategies for CCDS treatment.
Creatine is critical for the maintenance of energy homeostasis. A decrease in creatine levels in the central nervous system (CNS) leads to cerebral creatine deficiency syndrome (CCDS), referred to as neurodevelopmental disorders.1) CCDS is caused by functional mutations in creatine-synthetic enzymes or creatine transporter (CRT/SLC6A8).1,2) Mutations in SLC6A8 are the most frequent causes of CCDS and account for approximately 2% of X-linked intellectual disabilities.3–5) Creatine administration is insufficient for patients with CCDS with SLC6A8 mutations; however, it improves the symptoms of CCDS with mutations in creatine synthetic enzymes.1) To establish effective treatment strategies for CCDS with SLC6A8 mutations, normalization of mutated SLC6A8 has been proposed for creatine supplementation of target cells, such as neurons.6) Understanding the mechanisms underlying SLC6A8 dysfunction caused by mutations is useful for normalizing the mutated SLC6A8.
SLC6A8 consists of 12 transmembrane domains (TMs) and plays a vital role in the distribution of creatine in tissues, including brain tissue.3,7) SLC6A8 mediates creatine transport from the extracellular space to intracellular space across the plasma membrane, driven by the concentration gradient of sodium and chloride ions.3,8) Patients with functional SLC6A8 mutations show decreased creatine distribution and energy levels in tissues.1,2) A previous study hypothesized that impaired localization of SLC6A8 to the plasma membrane induces SLC6A8 dysfunction in missense mutations.9) Glycosylation on N192 and N197 of SLC6A8 is reportedly critical for functional expression.10) The c.1681G > C (p.G561R) mutation in SLC6A8 results in incomplete N-glycosylation and impaired localization to the plasma membrane.9) These studies suggest that SLC6A8 mislocalization due to abnormal glycosylation leads to SLC6A8 dysfunction. In addition, several missense SLC6A8 mutations have been reported in CCDS, as well as a few nonsense and frameshift mutations.5) However, the detailed mechanisms underlying the dysfunction of mutated SLC6A8, except for its mislocalization, are not yet fully understood.
In clinical studies, several patients with SLC6A8 mutations have been reported to show partial improvement of symptoms after the administration of creatine and its precursors, although this is not sufficient to overcome CCDS.1) The SLC6A8 mutations c.757G > C (p.G253R), c.859del (p.287fs), c.1006_1008del (p.336del), c.1067G > T (p.G356V), and c.1699T > C (p.S567P) are known.1) This study aimed to identify the dysfunctional mechanism(s) of SLC6A8 mutations by using in vitro overexpression systems. Considering the data obtained in this study, we sought to clarify the detailed mechanism underlying SLC6A8 dysfunction caused by the c.1699T > C mutation.
All chemicals used in this study were of analytical grade. Creatine hydrate [4-14C]- ([14C]creatine, 57.0 mCi/mmol) was purchased from Moravek Biochemicals (Brea, CA, U.S.A.).
Transport AnalysesTransport in Xenopus laevis oocytes was conducted as previously described and expressed as the oocyte/medium ratio (Eq. 1).11) The maximal uptake rate (Vmax) and Michaelis–Menten constant (Km) were estimated using Eq. 2 using a nonlinear least-squares regression analysis program (MULTI).12) In Eq. 2, V and [S] represent the uptake rate and creatine concentration, respectively. Details are provided in the Supplementary Materials.
 | (1) |
 | (2) |
Immunoblotting and immunocytochemistry were conducted as previously described11,13) and are provided in Supplementary Materials. For comparison, cells expressing SLC6A8 wild type (WT) or mutants were treated using the same procedures and images were captured under the same conditions.
Statistical AnalysesAll data are presented as mean ± standard deviation (S.D.). Statistical analyses of two groups and more than two groups were performed using the unpaired two-tailed Student’s t-test and one-way ANOVA followed by Dunnett’s test, respectively.
Creatine transport activity was evaluated using five human SLC6A8 mutants (c.757G > C, c.859del, c.1006_1008del, c.1067G > T and c.1699T > C) expressed in oocytes. [14C]Creatine uptake into SLC6A8 WT-expressing oocytes showed a 321-fold increase compared to that in water-injected oocytes (Fig. 1A). Because water-injected oocytes hardly exhibited endogenous creatine uptake, creatine accumulation in SLC6A8-expressing oocytes was attributed to SLC6A8-mediated transport. Among the five mutations, the c.1699T > C mutant exhibited residual transport activity of SLC6A8, whereas the other mutants exhibited minimal creatine transport activity (Fig. 1B). Uptake in SLC6A8 WT and c.1699T > C mutants decreased by >90% in the presence of 1 mM unlabeled creatine (Figs. 1C, D). These results indicate that SLC6A8 with the c.1699T > C mutation retained creatine transport activity. Further studies were performed to elucidate the detailed mechanism underlying the c.1699T > C mutation.
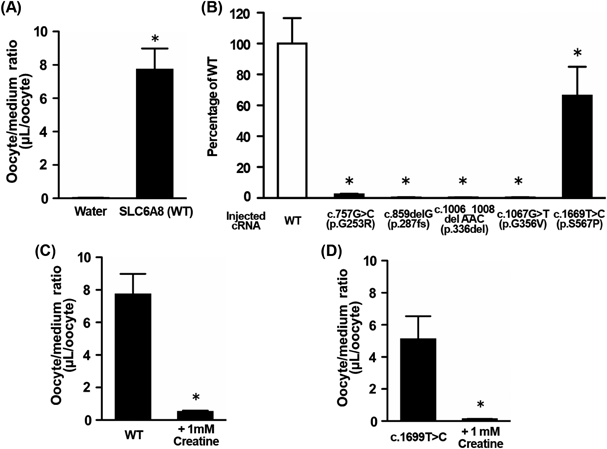
(A) Uptake of [14C]creatine by oocytes injected with water or SLC6A8 WT cRNA was measured for 60 min at 20 °C. Each column represents the mean ± S.D. (n = 7). * p < 0.01, significantly different from water-injected oocytes. (B) Uptake of [14C]creatine by WT or mutated SLC6A8-expressing oocytes was measured for 60 min at 20 °C. Each column represents the mean ± S.D. (n = 7). * p < 0.01, significantly different from SLC6A8 WT cRNA-injected oocytes. (C, D) Uptake of [14C]creatine by WT (C) or c.1699T > C mutant (D) was measured in the presence or absence of 1 mM unlabeled creatine. Each column represents the mean ± S.D. (n = 7). * p < 0.01, significantly different from SLC6A8 WT- or c1699T > C mutant-expressing oocytes.
To elucidate the details of the altered creatine transport in the c.1699T > C mutation, in vitro studies were performed using Flag- and Xpress-tagged SLC6A8-overexressing mammalian cells and Xenopus oocytes. [14C]Creatine uptake by SLC6A8 WT was not altered by the fusion tag (Supplementary Fig. S1). In immunoblot analyses, mature SLC6A8 at approximately 68 kDa9) was detected in SLC6A8 WT and c.1699T > C-expressing oocytes, which is consistent with the reported size of mature SLC6A8. However, dimers or trimers of immature SLC6A8, which are reportedly localized at the intracellular membrane, were also observed at approximately 150 kDa9) (Fig. 2A). A comparison of the intensity of the band derived from mature SLC6A8 between the samples suggested that the protein level of mature SLC6A8 protein was not changed in the c.1699T > C mutant compared to that in the WT (Fig. 2A). Furthermore, fluorescence derived from the tag was detected on the plasma membrane in SLC6A8 WT- and c.1699T > C mutant-expressing oocytes and HEK293 cells (Figs. 2B, C), indicating that SLC6A8 with the c.1699T > C mutation was localized to the plasma membrane, as well as SLC6A8 WT. In the immunoblotting, nonspecific bands were detected in water, SLC6A8 WT and c.1699T > C mutant. However, the signal was not observed in the sample of water-injected oocytes in immunostaining. Thus, detected signals in immunostaining were derived from tagged-SLC6A8, at least. Although immature SLC6A8 in intracellular compartments was not observed in immunostaining images, immature SLC6A8 was dominantly detected in the blotting. The signal of mislocalization mutation such as c.1681G > C was reported to be detected intracellularly.9) However, c.1699T > C mutant can be localized on the plasma membrane. To determine the localized level of the c.1699T > C-mutated SLC6A8 on the plasma membrane, quantitative evaluation of the protein localization might be needed as further studies. Nevertheless, the plasma membrane localization of SLC6A8 was qualitatively evaluated in the immunostaining. The c.1699T > C mutation causes a substitution of serine with proline at amino acid 567, which lies in the TM domain. Amino acid substitution in the TM domain can induce misfolding of membrane proteins and abnormal accumulation in the endoplasmic reticulum (ER).14) However, the present study showed no alteration in plasma membrane localization in the c.1699T > C mutant, suggesting that the alteration of protein expression and localization with translational and post-translational modifications is not mainly involved in the dysfunction of creatine transport by the c.1699T > C mutation.

(A) Immunoblot analysis of SLC6A8 with c.1699T > C mutation in oocytes. The crude membrane fraction of the oocytes (50 µg/lane) was separated using electrophoresis on a polyacrylamide gel. The arrowhead indicates mature SLC6A8 proteins. (B) Immunocytochemical analysis of SLC6A8 with c.1699T > C mutation in HEK293 cells. WT (B1) or c.1699T > C mutation (B2) of SLC6A8 was transiently expressed in HEK293 cells and stained with anti-Flag tag antibodies. The arrowheads indicate tagged SLC6A8 localized on the plasma membrane. Scale bar: 10 µm. (C) Protein localization of mutated SLC6A8 in oocytes. The sections of oocytes injected with SLC6A8 WT cRNA (C1), SLC6A8 with c.1699T > C mutation cRNA (C2), SLC6A8 with c.428A > T mutation cRNA (C3), or water (C4) were stained with anti-Xpress tag antibodies. Arrowheads indicate tagged SLC6A8 localized on the plasma membrane of oocytes. Scale bar: 50 µm. (D) Concentration-dependent uptake of creatine by WT-, c.1699T > C mutant-, or c.428A > T mutant-expressing oocytes. The uptake was performed for 60 min at 20 °C. A1 and A2 represent the Michaelis–Menten plot and the Eadie–Scatchard plot, respectively. Each point represents the mean ± S.D. (n = 3–5). (E) The structure model of SLC6A8 (UniProt ID: P48029) from the AlphaFold protein structure database.15,16) Dashed lines indicate hydrogen bonds. E2 is a different angle from E1.
In the concentration-dependent uptake of creatine in SLC6A8-expressing oocytes, Vmax in the c.1699T > C mutant was 117 µmol/(h·oocyte), which was 41.7% of that in the WT (Fig. 2D, Table 1). In contrast, the Km of WT and c.1699T > C mutant were obtained as 65.4 and 58.7 µM, respectively, and these values were not significantly changed (Fig. 2D, Table 1), suggesting similar recognition for creatine in both WT and c.1699T > C mutation. Although the degree of reduction in transport activity of c.1699T > C mutant was different between Fig. 1B and Table 1, the value in physiological condition is appropriate for considering the transport activity. In c.1699T > C mutant, the velocity of creatine transport calculated from the Michaelis–Menten equation was reduced by approx. 45% in the plasma concentration of creatine (25–100 µM). Thus, transport activity of c.1699T > C mutant is considered to remain approx. 45% in the physiological condition. The ratio of creatine level in the brain parenchyma in the patient with c.1699T > C mutation to normal was reported to be 0.36 and this ratio is a little higher than that of the other reported mutant (20–30%).1) In contrast, the IQ level in the patient with c.1699T > C mutation was reported to be moderate (IQ 35–50). Considering our results of the transport study, creatine seems to be provided into the brain in the patient with c.1699T > C mutation. However, as the patient with c.1699T > C mutation shows developmental disorder,1) the creatine amount is considered to be insufficient. Although the c.1699T > C mutant was normally localized at the plasma membrane and the Km of the c.1699T > C mutant was similar to that of the WT, the Vmax was decreased in the c.1699T > C mutant. Therefore, another mechanism apart from localization and substrate recognition is involved in SLC6A8 dysfunction due to the c.1699T > C mutation.
| Vmax (µmol/[h·oocyte]) | Km (µM) | |
|---|---|---|
| WT | 278 ± 35 | 65.4 ± 23.4 |
| c.1699T > C (p.S567P) | 117 ± 20* | 58.7 ± 15.7 |
| c.428A > T (p.Y143F) | 121 ± 16* | 72.9 ± 27.9 |
Vmax and Km values were obtained from uptake studies using SLC6A8 WT, c.1699T > C mutant, or c.428A > T mutant-expressing oocytes (Fig. 2D). As creatine accumulated minimally in water-injected oocytes and the accumulation was approximately 100-fold more in SLC6A8-expressing oocytes compared to that in water-injected oocytes, creatine accumulation in SLC6A8-expressing oocytes was attributed to SLC6A8-mediated transport. Kinetic parameters were obtained from the results of uptake in SLC6A8-expressing oocytes. Each value represents the mean ± S.D. (n = 3–5). * p < 0.01, significantly different from the WT.
To understand the mechanism of the hypoactivity in plasma membrane-localized c.1699T > C mutant, the predicted SLC6A8 structure (UniProt ID: P48029) was obtained from the AlphaFold protein structure database.15,16) In this model, hydrogen bonds were observed between the side groups of S567 in TM-12 and Y143 in TM-3 (Fig. 2E). Transport studies were performed using oocytes expressing the c.428A > T (p.Y143F) mutant, an artificial mutant predicted to abolish the hydrogen bond between S567 and Y143. On the plasma membrane of the oocytes, the SLC6A8 protein with the c.428A > T mutation and SLC6A8 WT were detected (Fig. 2C). Furthermore, Vmax for creatine uptake by Xenopus oocytes of SLC6A8 with the c.428A > T mutation was 43.5% of that in WT. In contrast, the Km value of the c.428A > T mutant was similar to that of the WT (Fig. 2D, Table 1). The kinetic parameters of the c.428A > T and c.1699T > C mutants were consistent (Fig. 2D, Table 1). Thus, these results show the possibility that S567 interacts intramolecularly with Y143 and that this interaction is essential for the normal transport activity of SLC6A8.
Among the SLC6A family transporters, TM-1 and TM-3 are critical for substrate recognition, whereas the contribution of TM-12 to substrate recognition is minor.17,18) In a homology model of SLC6A8, TM-1 of SLC6A8 was reported to be involved in polar interactions with the carboxylic group of creatine in the best docking pose.18) The C144 residue, which is located in TM-3, has been reported to form an ionic interaction with the guanidino group of creatine.18) Although Y143 lies next to C144, Y143 is located opposite to C144 (Fig. 2E2). In addition, the functional study showed no difference in Km values between SLC6A8 WT, c.1699T > C mutation, and c.428A > T mutation (Fig. 2D, Table 1). Thus, changes in creatine recognition are considered to be minor factors in SLC6A8 dysfunction caused by the c.1699T > C mutation. In contrast, Vmax was decreased in the c.1699T > C mutation compared to that in the WT (Fig. 2D, Table 1). The decrease in Vmax was considered to be induced after creatine bound to SLC6A8, as Km values did not change. Regarding SLC22A4, the reduction in Vmax without affecting Km is reportedly caused by conformational changes necessary for substrate transport.19) Therefore, the c.1699T > C mutation may cause conformational changes in SLC6A8 after substrate binding.
In conclusion, the present study proposes a novel mechanism for SLC6A8 dysfunction caused by the c.1699T > C missense mutation. SLC6A8 with the c.1699T > C mutation exhibited reduced transport activity, whereas its localization to the plasma membrane was similar to that of the WT, suggesting efficiency of creatine administration in part of CCDS patients. Our study implies that the interaction between amino acid residues affects the SLC6A8-mediated creatine transport in dysfunctional mutations. Further studies on SLC6A8-mediated transport with double point mutant and the mechanisms of the other mutations will support our idea is important for building a treatment strategy of CCDS.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Nos. 20H03403 and 23H02647) and JST SPRING (Grant No. JPMJSP2145).
RJ designed the study. RJ and MS collected data. RJ performed data analysis. KH coordinated the study. RJ drafted the manuscript, and YT, SA, MT, and KH revised it. All the authors have read and approved the final manuscript.
The authors declare no conflict of interest.
This article contains supplementary materials.