Abstract
Omeprazole, a gastric acid pump inhibitor, is repeatedly administered and is oxidatively metabolized mainly by polymorphic cytochrome P450 2C19. The prescribed dosage of omeprazole was discontinued or reduced in 47 of the 135 patients who received omeprazole alone in this survey, as recorded in the Japanese Adverse Drug Event Report database. The days to onset of omeprazole-related disorders were 3–4 d (median) and 16 d for intravenous 20–40 mg and oral 20 mg daily doses, respectively, in 34 patients for whom relevant data were available. The maximum plasma concentration of omeprazole was pharmacokinetically modeled after a single oral 40-mg dose in P450 2C19-defective poor metabolizers and was 2.4-fold higher than that in extensive metabolizers. The modeled area under the hepatic concentration curves of omeprazole in P450 2C19 poor metabolizers after virtual daily 40-mg doses for 7 d was 5.2-fold higher than that in the extensive metabolizers. Omeprazole-induced P450 2C19 (approx. 2-fold), resulting in increased hepatic intrinsic clearance in repeated doses, was considered after the second day. Virtual plasma/hepatic exposure estimated using pharmacokinetic modeling in subjects with P450 2C19 poor metabolizers indicated that these exposure levels virtually estimated could be one of causal factors for unexpected hepatic disorders induced by prescribed omeprazole, such as those resulting from drug interactions with repeatedly co-administered medicines.
INTRODUCTION
Interindividual differences in various drug-metabolizing enzymes would affect drug pharmacokinetics and impair therapeutic responses. The Japanese Adverse Drug Event Report (JADER) database,1) is generally used to mine rare adverse events that might not be noticed or reported in clinical trials and to study the time to the onset of drug-associated adverse events.2–4) Although various factors may contribute to drug-dependent adverse events, individual variations in the in vivo cytochrome P450 (P450)-dependent intrinsic clearance of drugs could be one of causal factors for the adverse events associated with the prescription of a single drug. Omeprazole, a gastric acid pump inhibitor used for the treatment of peptic ulcers, is administered repeatedly, often for a period of weeks.5) The pharmacokinetics and oxidation rates of omeprazole, a typical human P450 2C19 probe substrate, are associated with polymorphic human P450 2C19 phenotypes.6,7) A clear induction (from approx. 5- to 60-fold) of human P450 1A2 by omeprazole was evident in vitro in experiments with hepatocytes, in contrast to the contradictory reported induction results of human P450 2C9/2C19 by rifampicin.8) Metabolic activation of omeprazole by P450 3A4 to a reactive quinone imine metabolite has been reported.9) Modification of P450 2C19 expression by omeprazole was recently demonstrated10) using stable and reproducibly generated human HepaSH cells obtained from engrafted livers in immunodeficient humanized-liver mice.11)
The purpose of the present study was to evaluate the virtual plasma/hepatic exposures to omeprazole in P450 2C19 poor metabolizer subjects (approx. 20% in the Japanese population12)) using physiologically based pharmacokinetic (PBPK) modeling, as described in our related reports.13–16) Adverse events reported in the JADER database associated with the prescription of a single drug13–16) were focused on current and recent studies. In the present study, individual differences in hepatic exposure to omeprazole, modeled after virtual oral administration, were estimated in combination with inducible P450 2C19 wild-type and defective alleles. Therefore, we focused on omeprazole and estimated the virtual plasma and hepatic exposures of P450 2C19 poor metabolizers using a simplified PBPK modeling technique.
MATERIALS AND METHODS
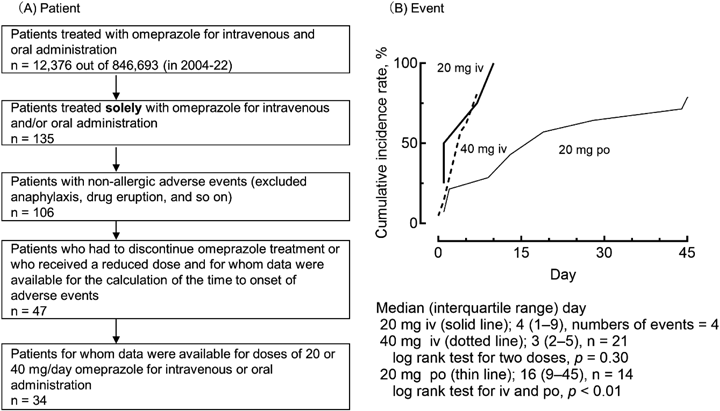
Items in the JADER database recorded between April 2004 and December 2022 were surveyed for adverse events in patients treated with omeprazole alone in a manner similar to that described previously.13–16) The number of days to onset was calculated from the date of occurrence of an adverse event for 20 or 40 mg omeprazole and the date of treatment initiation (recorded as drug regimen “discontinued” or “reduced”). The cumulative incidence and adverse events related to hepatic disorders were also analyzed (Table 1, Fig. 1).
Table 1. Adverse Events (
n = 124) Were Associated with Omeprazole Therapy in 106 Subjects Who Received No Other Prescription Drugs and Who Had to Discontinue Therapy or Receive a Reduced Dose
| Adverse event | Number of cases | Percentage (%) |
|---|
| Total | 124 | 100 |
| Tubulointerstitial nephritis and uveitis syndrome | 7 | 5 |
| Liver disorder | 6 | 4 |
| Tubulointerstitial nephritis | 6 | 4 |
| Agranulocytosis | 6 | 4 |
| Drug-induced liver injury | 6 | 4 |
| Inappropriate antidiuretic hormone secretion | 5 | 3 |
| Platelet count decreased | 4 | 3 |
| White blood cell count decreased | 4 | 3 |
| Aspartate aminotransferase increased | 3 | 2 |
| Hepatitis fulminant | 2 | 1 |
| Hepatic function abnormal | 2 | 1 |
| Jaundice | 2 | 1 |
| Hepatic failure | 1 | 1 |
| Hepatitis acute | 1 | 1 |
| Alanine aminotransferase increased | 1 | 1 |
| Others | 68 | 55 |
The data were obtained from the JADER database (Fig. 1). Some subjects experienced multiple adverse events. Allergy-related adverse events such as anaphylaxis, toxic epidermal necrolysis, and drug eruptions were excluded.
In this study, plasma concentration-versus-time data for omeprazole, previously reported after oral administration to Japanese subjects,17) were used to determine the pharmacokinetics of P450 2C19 extensive metabolizers (wild-type 2C19*1 homozygotes). The absorption rate constant (ka), volume of systemic circulation (V1), and in vivo hepatic intrinsic clearance (CLh,int) were estimated for a simplified human PBPK model as described previously13–16) and are briefly outlined in the footnote of Table 2. P450 2C19 extensive metabolizers (harboring homozygous 2C19*1) and defective poor metabolizers (harboring 2C19*2 or 2C19*3 alleles) were used to estimate in vivo hepatic intrinsic clearance values in the subjects. As the reported in vitro fraction metabolized omeprazole by P450 2C19 is 0.68,18) the estimated in vivo P450 2C19-dependent hepatic intrinsic clearance value for poor metabolizers was reduced to 0.32-fold. In humans, omeprazole can induce P450 2C19, which is involved in its 5-hydroxylation by approximately 2-fold.10) The P450 2C19-dependent intrinsic hepatic clearance was multiplied by 2 to model the repeated administration of omeprazole to P450 2C19 extensive metabolizers after the second day in this study. Plasma and liver concentrations estimated using PBPK models in virtual P450 2C19 extensive and poor metabolizer subjects are summarized in Table 3.
Table 2. Chemical Properties and Calculated Parameters for PBPK Modeling of Omeprazole Based on Reported Pharmacokinetic Data
| Parameter | Abbreviation (unit) | Value |
|---|
| Octanol–water partition coefficient | log P | 2.57 |
| Plasma unbound fraction | fu,p | 0.0430 |
| Blood–plasma concentration ratio | Rb | 0.762 |
| Liver (kidney)–plasma concentration ratio | Kp,h/Kp,r | 3.01 |
| Fraction absorbed × intestinal availability | Fa·Fg | 0.485 |
| Absorption rate constant | ka (1/h) | 0.530 ± 0.180a) |
| Volume of systemic circulation | V1 (L) | 23.4 ± 6.4a) |
| Hepatic intrinsic clearance | CLh,int (L/h) | 316 ± 6a) |
| Hepatic clearance | CLh (L/h) | 11.9 |
| Renal clearance | CLr (L/h) | 0.1 |
| Maximum concentration in plasma | Cmax (ng/mL) | 276 (0.773)b) |
| Area under the concentration curve from 0 to 8 h in plasma | AUC8 (ng h/mL) | 1310 (1.19)b) |
The acid dissociation constant (pKa), plasma unbound fraction (fu,p), and octanol–water partition coefficient (log P) values were obtained by in silico estimation using ACD/Percepta, Simcyp, and ChemDraw packages, respectively. The liver (kidney)-to-plasma concentration ratios (Kp,h/Kp,r) and the blood-to-plasma concentration ratio (Rb) were calculated from the fu,p, and log P values as follows20):

a) Data from fitting estimations (mean ± standard deviation) were obtained from reported pharmacokinetic data in six Japanese subjects17) with 1% renal elimination.21) The following set of differential equations was solved for the amounts and concentrations:

where Xg, Vh, Vr, Ch, Cr, and Cb are the amount of drug in the gut compartment; liver and kidney volumes; and hepatic, renal, and blood substrate concentrations, respectively.22) Vh, Vr, and Qh/Qr are the liver (1.5 L) and kidney (0.28 L) volumes and the blood flow rates of the systemic circulation to the hepatic/renal compartments (96.6 L/h) in humans (70 kg body weight).22) b) PBPK model outputs after virtual oral administration of 40 mg omeprazole. The values in parentheses are the ratios to the observed values. Cmax, maximum concentration; AUC8, area under the concentration curve from 0 to 8 h. Because the in vitro fraction metabolized of omeprazole by P450 2C19 has been reported to be 0.68, the estimated in vivo hepatic intrinsic clearance for P450 2C19 poor metabolizers was set at 101 L/h (i.e., 316 L/h ×0.32).
Table 3. Estimated Plasma and Liver
Cmax and
AUC168 Values Obtained Using Human PBPK Models after Virtual Oral Doses of 40 mg Omeprazole Daily for 7 d in Virtual P450 2C19 Extensive and Poor Metabolizer Subjects
| Plasma/liver | P450 2C19 phenotype | Cmax, µg/mL or µg/g | AUC168, µg h/mL or µg h/g |
|---|
| Plasma | Extensive metabolizer | 0.20 (1.0) | 6.0 (1.0) |
| Poor metabolizer | 0.47 (2.4) | 31 (5.2) |
| Liver | Extensive metabolizer | 0.62 (3.1) | 18 (3.0) |
| Poor metabolizer | 1.4 (7.0) | 94 (16) |
The modified in vivo hepatic intrinsic clearance values were used for the two genotypes (Table 2). In humans, omeprazole can induce (approx. 2-fold) P450 2C19, which is involved in its 5-hydroxylation.10) Cmax, maximum concentration; AUC168, area under the concentration curve from 0 to 168 h. The numbers in parentheses indicate the fold increase in plasma Cmax or AUC168 values with respect to P450 2C19 extensive metabolizers.
RESULTS
Of the 12376 patients in the JADER database treated with intravenous or oral omeprazole, 135 were treated with omeprazole alone (Fig. 1A). Reduced or discontinued dosages were recorded in 47 of these patients, who experienced 124 adverse events in total, excluding allergy-related adverse events such as anaphylaxis, toxic epidermal necrolysis, and drug eruptions (Table 1). The apparent incidence was high (35%). Among the 34 patients with data on the timing of adverse events, the onset of adverse events was 4 (1–9) and 3 (2–5) days (median [interquartile range] number of days) for intravenous 20-mg and 40-mg daily doses, respectively, and 16 (9–45) days for oral 20-mg daily doses of omeprazole, respectively (Fig. 1B). There was a significant difference in the number of days to onset between the omeprazole administration groups (Fig. 1B). No significant differences were observed between the two intravenously administered dosages, suggesting that factors other than dosage were responsible for the adverse events.
Using the previously reported plasma concentration plots of omeprazole after an oral dose of 40 mg in P450 2C19 extensive metabolizers (homozygous wild-type 2C19*1),17) the virtual time-dependent plasma and hepatic concentration curves are shown in Fig. 2A. Despite various factors for unexpected drug-dependent adverse events, the model output results for P450 2C19-defective poor metabolizer subjects (harboring the defective alleles 2C19*2 or 2C19*3) are shown in Fig. 2B, using reduced in vivo hepatic intrinsic clearance values relative to wild-type 2C19*1, as described in the footnote of Table 2. The P450 2C19-dependent intrinsic hepatic clearance was multiplied by 2 when modeling repeated administrations of omeprazole to P450 2C19 extensive metabolizers after the second day, to account for autoinduced metabolism.10) After repeated virtual doses of 40 mg omeprazole for 7 d, the maximum plasma concentration (Cmax) and the area under the concentration curve from 0 to 168 h (AUC168) in P450 2C19 poor metabolizers were 2.4- and 5.2-fold higher, respectively, than those in P450 2C19 extensive metabolizers (Figs. 2C, D, Table 3). The plasma and hepatic Cmax values generated by the PBPK models were 0.20 µg/mL and 0.62 µg/g, respectively, in P450 2C19 extensive metabolizers. Plasma and hepatic Cmax concentrations increased to 0.47 µg/mL and 1.4 µg/g, respectively, in P450 2C19 poor metabolizers.
DISCUSSION
Although various factors are responsible for unexpected adverse events caused by drugs, we speculate that increased plasma and/or hepatic exposure resulting from polymorphisms in the responsible enzyme gene may also induce hepatic adverse events when drugs are prescribed alone. This may result in dose reduction or discontinuation of the prescribed drug, as also happens when concomitant therapy causes drug-metabolizing enzyme inhibition, resulting in a variety of drug–drug interactions.9) Previously, we estimated higher plasma and liver concentrations13–16) of the model drugs celecoxib, diclofenac, and fluvastatin in patients with impaired P450 2C9.3, atomoxetine in patients with impaired P450 2D6.10, and atorvastatin in patients with impaired P450 3A4.16, which are all caused by amino acid substitutions in responsive P450 isoforms. In the present study, P450 2C19-dependent hepatic intrinsic clearance values for omeprazole and loss of function in poor metabolizers were used to estimate the hepatic and plasma drug concentrations after virtual oral administration. Considering the in vitro or in vivo contributions of the inducible enzyme responsible for the omeprazole oxidation reaction, the in vivo loss of enzyme function in poor metabolizers was considered in this study. The factors causing adverse events in patients administered omeprazole alone could be related to tubulointerstitial nephritis or liver disorders (Table 1), which account for up to 18% of liver-related disorders. This value is similar to that of the Japanese P450 2C19 poor metabolizers (approx. 20%). We speculate that genetic polymorphisms of CYP2C19 likely constitute another contributing factor to adverse events in patients prescribed omeprazole alone, resulting in high exposure to its possible metabolic activation by other enzyme P450 3A4.9) In our separate JADER analysis, rhabdomyolysis was also associated with omeprazole co-administration, even without concomitant statins or fibrates, indicating a large difference in time-to-onset days (17.5–196 d) for oral omeprazole treatment.19)
The high plasma/hepatic exposure to omeprazole in this genetically distinct population (Table 3) indicates that defective P450 2C19 activity could be a causal factor for adverse effects (Table 1, Fig. 1). In future studies, establishing a relationship between adverse events and actual drug exposure in patients will be the most informative. Poor metabolizers with defective P450 2C19 may experience modified enzymatic function similar to that resulting from the co-administration of omeprazole and other prescribed medicines. In conclusion, the current results suggest the importance of considering the effects of possible genetic deficiencies on the expression of the enzyme responsible for metabolism, as well as the effects of P450 2C19 induction as a modifier of pharmacokinetics during repeated omeprazole administration. In Japan, the package inserts and interview form for original product for omeprazole have warnings about the risk of increasing plasma concentrations and potentiating effects, respectively, in patients with defective P450 2C19 poor metabolizers. However, this information is missing from Japanese generics. This situation appears to be different from that of celecoxib.14) The accumulation of pharmacogenomic information for drug package inserts in clinical practice in Japan is needed to confirm the present pharmacogenetic hypothesis regarding adverse events related to repeatedly administered drugs such as omeprazole.
Acknowledgments
The authors thank Hina Nakano, Haruka Nishimura, Atsuo Arai, Tsubasa Sasaki, and Manato Hosoi for their assistance. This work was supported in part by the Japan Agency for Medical Research and Development under Grant No. 24mk0101253h0103 and by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research 23K06217 and 23K14393. We are also grateful to David Smallbones for copyediting a draft of this article.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Tsuchiya M, Obara T, Sakai T, Nomura K, Takamura C, Mano N. Quality evaluation of the Japanese Adverse Drug Event Report database (JADER). Pharmacoepidemiol. Drug Saf., 29, 173–181 (2020).
- 2) Ohyama K, Hirakawa K, Sasazaki K, Tanaka H, Hori Y, Takeuchi H. Time-to-onset of diabetes with everolimus use: analysis of a spontaneous reporting system database. Pharmazie, 76, 515–518 (2021).
- 3) Ohyama K, Tanaka H, Hori Y. Effect of concomitant drug use on the onset and exacerbation of diabetes mellitus in everolimus-treated cancer. J. Pharm. Pharm. Sci., 25, 245–252 (2022).
- 4) Ohyama K, Akiyama S, Iida M, Hori Y. Association of torsade de pointes and QT prolongation with antifungal triazoles: analysis using a pharmacovigilance database. In Vivo, 37, 2719–2725 (2023).
- 5) Furuta T, Shirai N, Sugimoto M, Ohashi K, Ishizaki T. Pharmacogenomics of proton pump inhibitors. Pharmacogenomics, 5, 181–202 (2004).
- 6) Turpault S, Brian W, Van Horn R, Santoni A, Poitiers F, Donazzolo Y, Boulenc X. Pharmacokinetic assessment of a five-probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br. J. Clin. Pharmacol., 68, 928–935 (2009).
- 7) Yamazaki H, Inoue K, Shaw PM, Checovich WJ, Guengerich FP, Shimada T. Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeprazole by human liver microsomes: effects of contents of these two forms in individual human samples. J. Pharmacol. Exp. Ther., 283, 434–442 (1997).
- 8) Yajima K, Uno Y, Murayama N, Uehara S, Shimizu M, Nakamura C, Iwasaki K, Utoh M, Yamazaki H. Evaluation of 23 lots of commercially available cryopreserved hepatocytes for induction assays of human cytochromes P450. Drug Metab. Dispos., 42, 867–871 (2014).
- 9) Zhao Y, Sun C, Su M, Shi J, Hu Z, Peng Y, Zheng J. Evidence for metabolic activation of omeprazole in vitro and in vivo. Chem. Res. Toxicol., 35, 1493–1502 (2022).
- 10) Uno Y, Uehara S, Ushirozako G, Murayama N, Suemizu H, Yamazaki H. Cytochrome P450 1A2 and 2C enzymes autoinduced by omeprazole in dog hepatocytes and human HepaRG and HepaSH cells are involved in omeprazole 5-hydroxylation and sulfoxidation. Xenobiotica, 53, 465–473 (2023).
- 11) Uehara S, Higuchi Y, Yoneda N, Ito R, Takahashi T, Murayama N, Yamazaki H, Murai K, Hikita H, Takehara T, Suemizu H. HepaSH cells: Experimental human hepatocytes with lesser inter-individual variation and more sustainable availability than primary human hepatocytes. Biochem. Biophys. Res. Commun., 663, 132–141 (2023).
- 12) Inoue K, Yamazaki H, Imiya K, Akasaka S, Guengerich FP, Shimada T. Relationship between CYP2C9 and 2C19 genotypes and tolbutamide methyl hydroxylation and S-mephenytoin 4′-hydroxylation activities in livers of Japanese and Caucasian populations. Pharmacogenetics, 7, 103–113 (1997).
- 13) Adachi K, Ohyama K, Tanaka Y, Sato T, Murayama N, Shimizu M, Saito Y, Yamazaki H. High hepatic and plasma exposures of atorvastatin in subjects harboring impaired cytochrome P450 3A4*16 modeled after virtual administrations and possibly associated with statin intolerance found in the Japanese adverse drug event report database. Drug Metab. Pharmacokinet., 49, 100486 (2023).
- 14) Adachi K, Ohyama K, Tanaka Y, Nakano H, Sato T, Murayama N, Shimizu M, Saito Y, Yamazaki H. Plasma and hepatic exposures of celecoxib and diclofenac prescribed alone in patients with cytochrome P450 2C9*3 modeled after virtual oral administrations and likely associated with adverse drug events reported in a Japanese database. Biol. Pharm. Bull., 46, 856–863 (2023).
- 15) Shimizu M, Uehara S, Ohyama K, Nishimura H, Tanaka Y, Saito Y, Suemizu H, Yoshida S, Yamazaki H. Pharmacokinetic models scaled-up from humanized-liver mouse data can account for drug monitoring results of atomoxetine and its 4-hydroxylated and N-demethylated metabolites in pediatric patients genotyped for cytochrome P450 2D6. Drug Metab. Dispos., 52, 35–43 (2024).
- 16) Adachi K, Ohyama K, Tanaka Y, Saito Y, Shimizu M, Yamazaki H. Modeled hepatic/plasma exposures of fluvastatin prescribed alone in subjects with impaired cytochrome P450 2C9*3 as one of possible determinant factors likely associated with hepatic toxicity reported in a Japanese adverse event database. Biol. Pharm. Bull., 47, 635–640 (2024).
- 17) Uno T, Niioka T, Hayakari M, Yasui-Furukori N, Sugawara K, Tateishi T. Absolute bioavailability and metabolism of omeprazole in relation to CYP2C19 genotypes following single intravenous and oral administrations. Eur. J. Clin. Pharmacol., 63, 143–149 (2007).
- 18) McGinnity DF, Parker AJ, Soars M, Riley RJ. Automated definition of the enzymology of drug oxidation by the major human drug metabolizing cytochrome P450s. Drug Metab. Dispos., 28, 1327–1334 (2000).
- 19) Ohyama K, Iida M, Akiyama S, Yamazaki H, Hori Y. Time-to-onset analysis of rhabdomyolysis due to different proton pump inhibitors using a pharmacovigilance database. In Vivo, 38, 1285–1291 (2024), DOI: 10.21873/invivo.13567.
- 20) Adachi K, Utsumi M, Sato T, Nakano H, Shimizu M, Yamazaki H. Modeled rat hepatic and plasma concentrations of chemicals after virtual administrations using two sets of in silico liver-to-plasma partition coefficients. Biol. Pharm. Bull., 46, 1316–1323 (2023).
- 21) Nakashima M, Kanamaru M, Hashimoto H, Takiguchi Y, Mizuno A, Kajiho T, Oka T, Matsuda Y. Phase I study of omeprazole—single-dose and multiple-dose studies. Jpn. J. Clin. Pharacol. Ther., 19, 667–679 (1998).
- 22) Kamiya Y, Handa K, Miura T, Ohori J, Kato A, Shimizu M, Kitajima M, Yamazaki H. Machine learning prediction of the three main input parameters of a simplified physiologically based pharmacokinetic model subsequently used to generate time-dependent plasma concentration data in humans after oral doses of 212 disparate chemicals. Biol. Pharm. Bull., 45, 124–128 (2022).