Abstract
Background:
Levels of triglycerides and free fatty acids (FFAs) are elevated in patients with diabetes and may contribute to endothelial dysfunction through renin-angiotensin system (RAS) activation and oxidative stress. The present study investigated how systemic FFA loading affected myocardial microcirculation during hyperemia via RAS.
Methods and Results:
Eight healthy men received candesartan, perindopril, or a placebo for 2 days in a double-blind crossover design, and then myocardial microcirculation during hyperemia induced by a 2-h infusion of lipid/heparin was assessed using dipyridamole stress-myocardial contrast echocardiography (MCE). Leukocyte activity and hemorheology were also assessed ex vivo using a microchannel flow analyzer, serum levels of oxidative stress markers, and IκB-α expression in mononuclear cells. Serum FFA elevation by the infusion of lipid/heparin significantly decreased myocardial capillary blood velocity and myocardial blood flow during hyperemia. Both candesartan and perindopril significantly prevented the FFA-induced decrease in capillary blood velocity and myocardial blood flow during hyperemia. Systemic FFA loading also caused an increase in the number of adherent leukocytes and prolonged the whole blood passage time. These effects were blocked completely by candesartan and partially by perindopril. Both agents prevented the FFA-induced enhancement of oxidative stress and IκB-α degradation in mononuclear cells.
Conclusions:
Both candesartan and perindopril can prevent FFA-induced myocardial microcirculatory dysfunction during hyperemia via modulation of leukocyte activation and microvascular endothelial function.
Free fatty acids (FFAs), which are non-esterified fatty acids that circulate in the bloodstream predominantly bound to albumin, comprise the main oxidative fuel for the myocardium and skeletal muscles. Elevated plasma levels of FFAs are frequently observed in patients with type 2 diabetes and metabolic syndrome,1
and are associated with insulin resistance, inflammation, vascular dysfunction, and increased risk of cardiovascular mortality.2,3
Increased plasma FFA levels after lipid/heparin infusion or a meal result in endothelial dysfunction in healthy subjects.4–9
As potentially potent signaling molecules to induce endothelial dysfunction and insulin resistance, FFAs have been implicated in the pathogenesis of type 2 diabetes and atherosclerosis.4–6
We previously reported7,8
that FFA elevation results in renin-angiotensin system (RAS) activation in human leukocytes, leading to leukocyte activation and subsequent endothelial dysfunction. Our results suggest that alteration of leukocyte function by FFAs is a fundamental mechanism involved in the regulation of vascular function through the RAS,7,8
as well as other relevant vasoactive substances and cytokines.7–9
Leukocytes are critical to modulating microvascular hemodynamics through transformation from resting to active states under conditions of inflammation or low shear stress,10–12
such as a high-fat diet13
and ischemia/reperfusion.14
Decreased leukocyte deformability and increased leukocyte adherence to postcapillary venular endothelium synergistically lead to increased capillary resistance, in part through increased blood viscosity.12
Intravenous lipid infusion increases myocardial capillary resistance during adenosine stress hyperemia in dogs without affecting myocardial blood volume.15
We postulated that FFA elevation impairs myocardial microcirculation through modifying myocardial capillary resistance and microvascular endothelial dysfunction caused by leukocyte activation, in part via the RAS in humans. We tested this hypothesis in healthy individuals exposed to FFA provocation using dipyridamole-stress myocardial contrast echocardiography (MCE), as well as ex vivo microvascular models and blood laboratory analyses.
Methods
Subjects
Eighteen healthy non-smoking, non-obese (body mass index 20–24 kg/m2) and normotensive men (aged 20–35 years) who were not taking regular medication provided written informed consent to participate in the present study. All routine physical examinations and standard laboratory test results were normal. The Ethics Committee of the University of the Ryukyus approved the study protocol.
Lipid/Heparin Infusion
The participants fasted overnight and abstained from alcohol or caffeine for at least 12 h before the study. They arrived at the laboratory at 09:00 hour and received a continuous intravenous infusion of fat emulsion (Intralipid 20%; Fresenius Kabi AB, Uppsala, Sweden) at 90 mL/h and heparin (Ajinomoto Co. Ltd. Tokyo, Japan) at 0.3 U kg−1
min−1
to increase their serum FFA concentrations, as previously described.7–9
Heparin co-infusion activates lipoprotein lipase and catalyzes triglyceride hydrolysis.
Measurement of Plasma FFA Concentrations
Blood samples were mixed with 50 μL paraoxon (diethyl p-nitrophenyl phosphate; Sigma Chemical Company, St. Louis, MO, USA) diluted to 0.04% in diethyl ether to prevent ex vivo lipolysis.16
Plasma samples obtained after immediate centrifugation at 4℃ were stored at −80℃ until enzymatic assay.
Myocardial Contrast Echocardiography (MCE)
Independent technicians otherwise uninvolved in our study performed MCE using an Aplio system (Toshiba, Tokyo, Japan) with 1.5 harmonic pulse rate subtraction imaging. The participants were assessed in the left decubitus position by MCE before and after receiving an intravenous injection of dipyridamole (0.56 mg/kg) to induce pharmacological hyperemia. A levovist contrast agent (concentration of 300 mg/mL; Schering Japan, Osaka, Japan) was continuously intravenously infused using an infusion device (Pulsar, Medrad Indianola, PA, USA) at a rate of 600 mg/min (2.0 mL/min). Electrocardiograph-triggered end-systolic intermittent myocardial microvascular images with a mechanical index of 1.1–1.2 were acquired in the apical four-chamber view at incremental pulse intervals (triggering intervals of 1, 2, 4, and 6 cardiac cycles).17
Three or four images were captured at each pulse interval. All cardiac images were analyzed using Volumap-445 software (YD Inc., Nara, Japan). Video intensity was measured over a large region-of-interest placed in the anteroseptal myocardium at one-third of the distance from the apex to the mitral annulus (Figure 1A). Video intensity data at each pulse interval were fit to the formula using ORIGIN 6.0 J software (Lightstone, Tokyo, Japan), as previously described:18,19
y=A (1−e−βt), where y is video intensity at time t, A is the plateau video intensity reflecting myocardial blood volume, and β is the rate constant reflecting erythrocyte velocity in the capillaries. Myocardial blood flow was calculated as the product of A and β (A×β).

We used a microchannel flow analyzer (HR200; Kowa Co. Ltd., Tokyo, Japan) as an ex vivo model of capillaries and arterioles to assess whole blood rheology and leukocyte activity, as previously described.8–10,12,20–22
Within 10 min of collecting blood into heparinized tubes, 0.1 mL of blood was drawn through BK 7-7-4.5 microchannels (Kikuchi Microtechnology Co., Ltd. Ibaraki, Japan) as an ex vivo capillary model (7854-parallel, 7×4.5-μm equivalent cross-section, 30 μm long) under a constant vacuum of 20 cm H2O (1.96 kPa). The time required for saline to pass through the microchannels was determined before each blood measurement for calibration. Microscopic motion images of blood passing through microchannels were monitored and stored on a computer system. When 0.08–0.10 mL of blood had exited the microchannel array, 5 fields were recorded, 5 still images were randomly selected for off-line analysis, and the numbers of adhesive or clumped leukocytes on the microchannel platform in these images were counted. Adhesive leukocytes were defined as static leukocytes with a clear surface border on still images.8–10
To determine the apparent relative viscosity of pre-capillary arteriolar whole blood and plasma, we measured the time required for 0.05 mL of whole blood and plasma to pass through BK 7-7-7 microchannels (7854-parallel, 7×7-μm equivalent cross-sections, 30 μm long; Kikuchi Microtechnology Co., Ltd. Ibaraki, Japan) under a constant vacuum of 30 cm H2O (2.94 kPa) after calibration according to the duration of saline passage through the channels. In a pilot study, microchannels became occluded at a frequency of <0.2% (by activated leukocytes or platelet aggregation) per 30 samples in 10 men even after lipid/heparin loading. Therefore, the time taken for whole blood and plasma to pass through the channels divided by that of saline might represent apparent relative viscosities at pre-capillary arterioles.
Measurement of Derivatives of Reactive Oxygen Metabolites in Serum
We measured hydroperoxide levels as serum levels of reactive oxygen metabolite (d-ROM) derivatives using a FREE Carpe Diem photometer (Diacron srl, Grosseto, Italy).23,24
The d-ROM test depends on a Fenton-like reaction to produce lipid peroxy and alkoxy radicals that in turn react with a chromogenic substrate.23
Assessment of IκB-α in Human Mononuclear Cells From Peripheral Blood
Human mononuclear cells were isolated from blood collected with LymphoprepTM
medium (AXIS-SHIELD PoC AS, Oslo, Norway), as described by the manufacturer. The mononuclear cell fraction was mixed with 2% dextran 60,000 (Wako Pure Chemical Industries Ltd., Osaka, Japan) in Hank’s balanced salt solution for 30 min at room temperature. Mononuclear cells collected by centrifugation at 300 g for 5 min were suspended in medium 199 (Invitrogen) and counted. A suspension of 2×106
cells on ice was portioned in microtubes. Trichloroacetic acid was added to a final volume of 10%, and the mixture was separated by centrifugation at 16,000 g for 5 min at 4℃. Pellets were washed once with ice-cold Hank’s balanced salt solution and stored at −30℃ until all samples were processed. Pellets were sonicated in 9 mol/L urea (Wako Pure Chemical Industries Ltd.) containing 2% Triton X-100 (ICN Biomedicals Inc., Solon, OH, USA) and 1% dithiothreitol (MP Biomedicals, Solon, OH, USA). The protein concentrations in the lysates were then assessed using a dye-binding protein assay (Dye Reagent Concentrate; Bio-Rad Laboratories, Hercules, CA, USA). An equal volume of 2× sodium dodecyl sulfate sample buffer was added to the lysates, and the mixtures were heated to 96℃ for 3 min. Equal amounts of proteins were electrophoretically resolved on sodium dodecyl sulfate-polyacrylamide gels (Bio-Rad Laboratories). The separated proteins were transferred to polyvinylidine difluoride membranes (Bio-Rad Laboratories) and then immunoblotted against anti-IκB-α rabbit polyclonal antibody (Cell Signaling Technology, Boston, MA, USA) or anti-actin goat polyclonal antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 4℃ overnight. Horseradish peroxidase-linked secondary antibodies (GE Healthcare, Little Chalfont, Buckinghamshire, UK) were applied to the membranes, and immunoreactive proteins were detected using ImmunoStar LD (Wako Pure Chemical Industries Ltd.) and Hyperfilm ECL (GE Healthcare). Signals on Hyperfilm were scanned into a computer, and signal density was analyzed using Igor Pro software (WaveMetrics, Portland, OR, USA).
Effect of Sera on Endothelial Nitric Oxide (NO) Synthase Phosphorylation in Endothelial Cells (ECs)
Sera separated from blood samples collected before and 2 h after the lipid/heparin infusion were incubated at 56℃ for 30 min to inactivate complement. Bovine aortic ECs (DS Pharma Biomedical, Osaka, Japan) were cultured to confluence in 3.5-cm dishes containing medium 199 supplemented with 20% fetal bovine serum (FBS). The medium was then changed to FBS-free medium 199, and the cells were incubated for 1 h. Medium 199 containing 50% serum from each blood sample was incubated with ECs for another 1 h. The ECs were washed twice with ice-cold PBS, denatured with 10% trichloroacetic acid, and then lysed with urea. Equal amounts of protein were subjected to immunoblotting to detect total endothelial NO synthase (eNOS) expression and phosphorylation at Ser-1179 (the phosphorylation site of activated eNOS that is equivalent to human Ser-1177). All antibodies were purchased from BD Transduction Laboratories.
Study Protocols
Protocol 1. Effects of Increased Serum FFA With or Without RAS Inhibition on Myocardial Microcirculation
Eight normotensive healthy men underwent this 4-day protocol, with intervals of at least 14 days between tests. Each of the men received a placebo, the angiotensin II receptor blocker (ARB), candesartan (16 mg once daily), or the angiotensin-converting enzyme (ACE) inhibitor, perindopril (4 mg once daily), at 28 and 4 h before the MCE examination on study days 2–4 in a double-blind, crossover design. The order of drug or placebo administration was randomized. All of the men were assessed by MCE after systemic infusion of saline/heparin for 120 min as a control experiment on study day 1 and of lipid/heparin for 120 min on study days 2–4. Blood pressure and pulse rate were serially monitored at 0, 60, and 120 min (TM-2541R; A&D Co. Ltd, Tokyo, Japan).
Protocol 2. Effects of Increased Serum FFA With or Without RAS Inhibition on Whole Blood Rheology, Leukocyte Activity, and IκB-α Expression in Mononuclear Cells Ten healthy men underwent this 4-day protocol with intervals of at least 14 days between tests. As in protocol 1, each man received a placebo, candesartan (16 mg) once daily, perindopril (4 mg once daily), or placebo at 28 and 4 h before the experiment on study days 2–4 in a double-blind crossover design. Blood samples were collected from all participants to assess whole blood rheology and leukocyte activity using the microchannel flow analyzer and to measure serum levels of d-ROMs as an oxidative stress marker before and 60 and 120 min after starting systemic infusions of saline/heparin (control experiment; study day 1) and lipid/heparin (study days 2–5). Blood pressure and pulse rate were serially monitored as in protocol 1. Blood samples were collected from 6 of the 10 participants to estimate IκB-α expression in mononuclear cells at each sampling point ex vivo. Serum was also separated from blood samples to estimate the effects of sera after the lipid/heparin infusion with or without RAS inhibition on eNOS phosphorylation in bovine ECs (n=3).
Statistical Analysis
Data were statistically analyzed using JMP 7.01 J software (SAS Institute, Cary, NC, USA) and presented as mean±SD unless otherwise indicated. Probability values of P<0.05 were considered to indicate statistical significance. Individual MCE parameters (A value, β value, and capillary blood flow) between drug or placebo administrations and infusions were compared using ANOVA followed by the Tukey-Kramer post-hoc test. The duration of whole blood passage and number of adhesive leukocytes were compared between drug or placebo administrations and infusions using repeated-measures ANOVA followed by the Tukey-Kramer post-hoc test. Serial changes in relative viscosity and dROM results were also analyzed by repeated-measures ANOVA.
Results
Blood pressure, heart rate, and hematocrit before lipid/heparin infusion did not differ across experimental days, and remained unchanged during lipid/heparin infusion (data not shown). Serum triglyceride and FFA levels increased from 67.3±63.8 to 281.3±136.6 mg/dL and from 0.45±0.20 to 1.66±0.25 mEq/L, respectively, after the 1-h infusion of lipid/heparin, and to 415.5±260.8 mg/dL and 1.90±0.50 mEq/L, respectively, after the 2-h infusion (protocol 2, n=8).
1. Effect of Elevated FFA Levels on Myocardial Microcirculation During Hyperemia and the Role of the RAS
The A- and β-values did not significantly differ across the administered drugs or placebo after lipid/heparin infusion without dipyridamole stress (data not shown). The infusion of lipid/heparin significantly decreased capillary blood velocity (β-value,
Figure 1A
and
B) and myocardial blood flow (A×β-value,
Figure 1C) during dipyridamole-induced hyperemia. Capillary blood volume (A value,
Figure 1D), however, remained unchanged by the infusion of lipid/heparin. Both candesartan and perindopril significantly prevented the FFA-induced decrease in capillary blood velocity and myocardial blood flow during hyperemia (Figure 1B,C).
2. Effect of Elevated FFA Levels on Hemorheology in Microvascular Models Ex Vivo
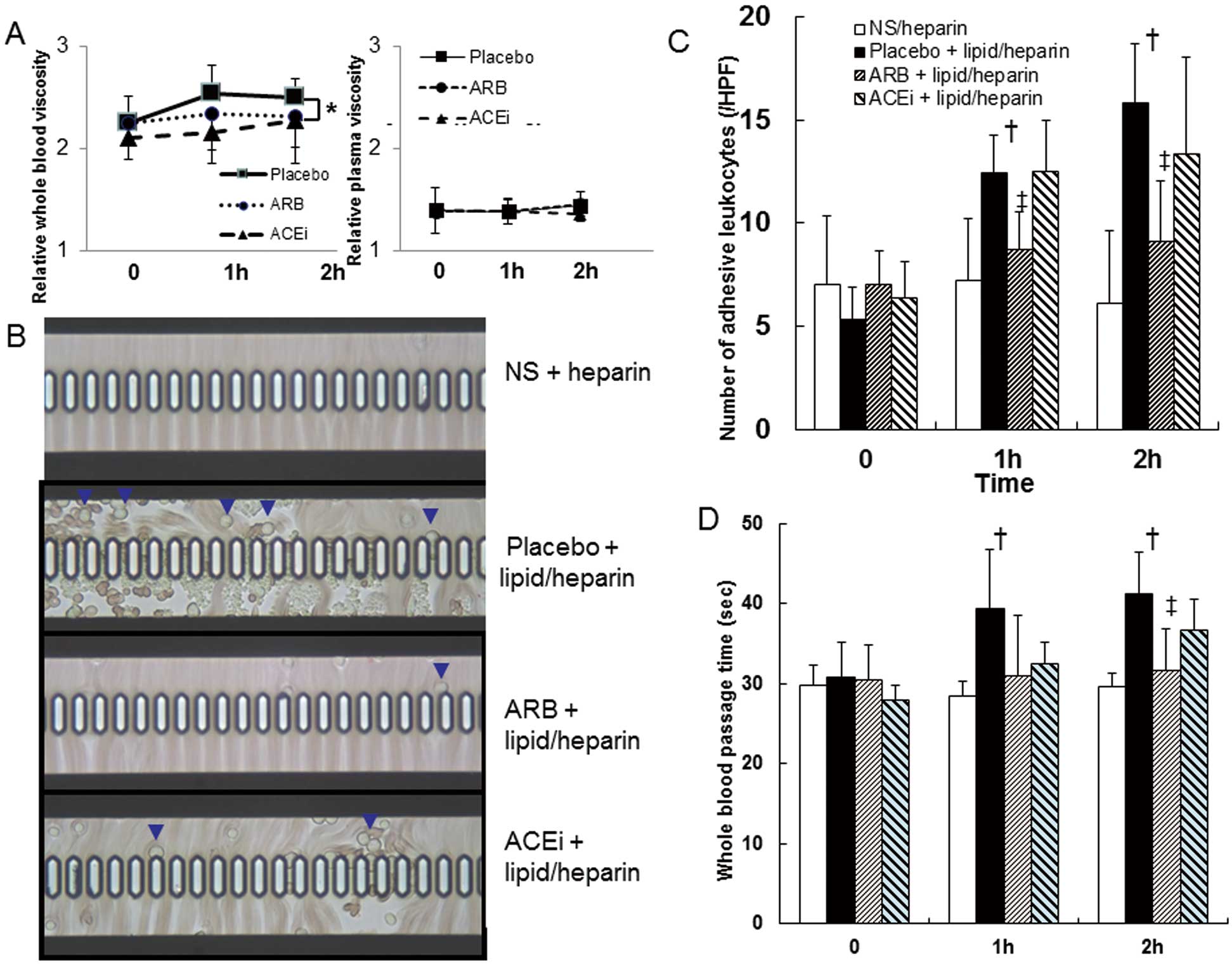
Figure 2A
shows serial changes in the apparent relative viscosity of whole blood and plasma in the ex vivo pre-capillary arteriolar model (BK7-7-7) before and during lipid/heparin loading. The relative whole blood viscosity increased from 2.24±0.25 to 2.54±0.27 at 1 h and to 2.49±0.18 at 2 h of lipid/heparin infused after placebo administration. Prior candesartan or perindopril treatment prevented the FFA-induced increase in whole blood viscosity (Figure 2A). Lipid/heparin infusion time-dependently increased the number of adherent or clumped leukocytes and the duration of whole blood passage in the ex vivo capillary model (BK7-7-4.5;
Figure 2B–D). Prior administration of candesartan significantly inhibited FFA-induced leukocyte activation at 1 and 2 h, whereas administration of perindopril did not (Figure 2C). Candesartan significantly inhibited the prolongation of whole blood passage by FFA at 2 h, whereas perindopril had a moderate but nonsignificant effect (Figure 2D).
Lipid/heparin infusion time-dependently increased serum levels of hydroperoxide measured using the d-ROM test (Figure 3A). Inhibiting the RAS with an ARB or ACE inhibitor partially prevented FFA-induced oxidative stress.
4. Effect of FFA Levels on IκB-α Expression in Mononuclear Cells and the Role of the RAS
Figure 3B
shows protein expression of IκB-α in mononuclear cells separated from peripheral blood at 0, 1, and 2 h after systemic loading of FFA. While saline/heparin infusion did not affect IκB-α expression, elevated FFA caused IκB-α degradation, which was prevented by the prior administration of either candesartan or perindopril.
5. Effects of Serum From Participants Infused Lipid/Heparin on eNOS Phosphorylation in ECs
While Ser-1179 phosphorylation was decreased by co-incubation of serum from participants administered placebo, serum from participants administered candesartan or perindopril enhanced Ser-1179 phosphorylation (Figure 4).
Discussion
In the present study, both candesartan and perindopril were able to prevent FFA-induced myocardial microcirculatory dysfunction during hyperemia via modulation of leukocyte activation and microvascular endothelial function. The elevation of plasma FFA originating from visceral adiposity in patients with metabolic syndrome and type 2 diabetes mellitus might result in endothelial, microvascular,25
and metabolic dysfunction such as insulin resistance.2,3
Liu et al have reported, using contrast ultrasonography, that FFA elevation by lipid/heparin infusion for 5 h blocked insulin-induced microvascular recruitment in both cardiac and skeletal muscle.26
Furthermore, the present study findings are the first evidence of FFA elevation damaging the human myocardial microcirculation during hyperemia. Capillary blood velocity (β) and myocardial blood flow (A×β) were reduced during dipyridamole-induced hyperemia, and activation of the RAS by elevated FFA contributed to impaired myocardial microcirculation, in part, via the modulation of leukocyte activity and microvascular endothelial function.
The present study findings support those of Rim et al, who showed that increasing doses of lipid emulsion do not affect myocardial blood volume (A), but increase myocardial vascular resistance by decreasing capillary velocity (β) during hyperemia in dogs.15
We postulated that the FFA-induced reduction in myocardial microvascular velocity is due to increased resistance in capillaries, because these vessels are responsible for 75% of the total microvascular resistance in maximally dilated coronary vasculature.27,28
In fact, the amount of time required for whole blood to pass through a microchannel array of capillaries with the same inner lumen diameter (7×4.5 μm), which reflects capillary resistance, was significantly prolonged after the lipid/heparin infusion. The numbers of adherent or clumped leukocytes in the ex vivo
capillary model were also significantly increased after lipid/heparin infusion. The present data are in line with a recent study that reported that lipid/heparin infusion induced an early protective endoplasmic reticulum stress response evidenced by activation of activating transcription factor 6 and phospho-inositol requiring kinase 1 in both leukocytes and ECs.29
Thus, leukocytes activated by FFA might transiently obstruct capillaries, adhere to the postcapillary venular endothelium, and consequently increase capillary resistance through increased leukocyte viscosity
in vivo.10–13,30
Indeed, we discovered that whole blood viscosity was increased in the pre-capillary model after lipid/heparin infusion, whereas FFA did not affect either plasma viscosity or hematocrit. Reduced flow and increased capillary viscosity might further activate leukocytes due to low venular shear stress and thus lead to a vicious circle of FFA-induced microvascular dysfunction and inflammation.11,12
Nishimura et al13
have notably observed that blood flow velocity is particularly sluggish in capillaries and postcapillary venules containing adherent leukocytes in adipose tissue of obese mice, whereas erythrocyte rheological properties remain unchanged. Leukocyte activation induced by FFA can explain these results, with which the present findings are consistent.
Activated leukocytes might also impair the endothelial function of microvessels, consisting of pre-capillary terminal arterioles, capillaries, and postcapillary venules, by releasing myeloperoxidase,8
reactive oxygen species, and cytokines. Substantial evidence suggests that enhanced oxidative stress is crucial to the modulation of vascular function by FFA.6,7,9,29,31–34
We found that FFA-induced myocardial microcirculation impairment was associated with elevated serum dROM levels and pro-inflammatory signaling activation in mononuclear cells.
The endothelial dysfunction and increased viscosity evoked by leukocyte activation might contribute to myocardial microcirculatory dysfunction, although direct effects of FFA on ECs cannot be excluded. Prior administration of either candesartan or perindopril prevented subsequent FFA-induced myocardial microcirculatory dysfunction during hyperemia, suggesting that the effects of FFA involve RAS activation. We assumed that enhanced angiotensin II production in mononuclear and polymorphonuclear cells by FFA8
causes leukocyte activation, which leads to increased viscosity and endothelial dysfunction of the microcirculation. In fact, prior administration of candesartan substantially inhibited FFA-induced leukocyte activation in the ex vivo capillary model and caused the subsequent increase in whole blood viscosity, presumably evoked via angiotensin II-induced signaling. By contrast, perindopril only partially prevented leukocyte activation in the ex vivo capillary model. One explanation for the different effects of the 2 drugs is that ACE is not the dominant enzyme required to form angiotensin II in leukocytes, especially in neutrophils.35,36
Therefore, ACE inhibitors alone cannot sufficiently block RAS-mediated signaling in neutrophils. The question remains as to how perindopril prevents the reduction of myocardial capillary blood flow during hyperemia.
The consequential production of NO in ECs is enhanced by ACE inhibitors via inactivation of bradykinin degradation. As NO from ECs might protect against leukocyte activation by increasing intra-leukocyte cGMP,11
perindopril should prevent leukocyte activation more efficiently via NO in vivo than ex vivo. Our hemorheology findings with this ACE inhibitor are consistent with a previous RCT in hypertensive patients with type 2 diabetes.37
These patients demonstrated higher whole-blood viscosity than controls, and whole-blood viscosity at shear rate gamma=128/s tended to decrease with enalapril.37
Candesartan and perindopril similarly inhibited the increase in serum dROM levels (representing enhanced oxidative stress) and IκB-α/NF-κB signaling activation in mononuclear cells. These results are consistent with the equivalent prevention of microcirculation impairment by these drugs and can be explained by the inhibition of angiotensin II signaling by candesartan and the enhanced production of NO in addition to a modest reduction in angiotensin II production by perindopril.38
We found that eNOS phosphorylation at Ser-1179 was modestly reduced after placebo and enhanced after either candesartan or perindopril in cultured ECs incubated with serum collected after a 2-h infusion of lipid/heparin. These results are consistent with previous findings
in vitro5
and in vivo,6–8,39
and suggest that angiotensin II/bradykinin are independent mechanisms involved in the prevention of FFA-induced microcirculatory dysfunction after the administration of candesartan and perindopril.
Study Limitations
First, we studied the effects of an ARB and an ACE inhibitor in healthy men before and after FFA provocation for only 2 h. Further investigations of long-term FFA provocation are warranted to extrapolate the present study results to obese patients, who have chronically elevated plasma FFA levels. Second, the duration of ACE inhibitor administration might have been relatively short. Third, erythrocyte fluidity was not assessed because of methodological issues, although FFA-induced oxidative stress might cause erythrocyte crenation and slight hemolysis.40,41
Finally, leukocyte behavior in the human myocardial microcirculation could not be visualized due to methodological limitations.
In conclusion, both candesartan and perindopril can prevent FFA-induced myocardial microcirculatory dysfunction during hyperemia via modulation of leukocyte activation and microvascular endothelial function, although their underlying mechanisms somewhat differ. Candesartan inhibited FFA-induced leukocyte activation, presumably via angiotensin II-induced signaling and reducing oxidative stress, while perindopril enhanced the bradykinin/NO pathway and decreased angiotensin II-induced signaling in mononuclear cells. Nevertheless, both drugs prevented the FFA-induced reduction in myocardial capillary blood velocity during hyperemia.
Clinical Perspective
Our results indicate that the RAS plays a role in the regulation of myocardial microvascular hemodynamics by modulating leukocyte activity and microvascular endothelial function under conditions of low-grade inflammation caused by FFA. We speculate that RAS inhibitors may confer protection against myocardial microcirculatory dysfunction in individuals with visceral fat obesity and patients with type II diabetes mellitus. Such patients are chronically exposed to high levels of circulating FFA, particularly in the clinical context of significant epicardial coronary artery stenosis.27
Acknowledgments
We thank Hiroyuki Kotsuka and Yuhki Hayakawa for assistance with the echocardiography.
Sources of Funding
This work was supported by Grants-in-Aid from the Ministry of Education, Science and Culture of Japan (16590439 to S.U.) and from the Vehicle Racing Commemorative Foundation to S.M. and T.Y.
Disclosure
The authors declare there are no conflicts of interest.
References
- 1.
Laws A, Hoen HM, Selby JV, Saad MF, Haffner SM, Howard BV, et al. Differences in insulin suppression of free fatty acid levels by gender and glucose tolerance status: Relation to plasma triglyceride and apolipoprotein B concentrations: Insulin Resistance Atherosclerosis Study (IRAS) Investigators. Arterioscler Thromb Vasc Biol 1997; 17: 64–71.
- 2.
Pilz S, Scharnagl H, Tiran B, Wellnitz B, Seelhorst U, Boehm BO, et al. Elevated plasma free fatty acids predict sudden cardiac death: A 6.85-year follow-up of 3315 patients after coronary angiography. Eur Heart J 2007; 28: 2763–2769.
- 3.
Pilz S, Scharnagl H, Tiran B, Seelhorst U, Wellnitz B, Bernhard O, et al. Free fatty acids are independently associated with all-cause and cardiovascular mortality in subjects with coronary artery disease. J Clin Endocrinol Metab 2006; 90: 3622–3628.
- 4.
Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Invest 1997; 100: 1230–1239.
- 5.
Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler Thromb Vasc Biol 2005; 25: 989–994.
- 6.
Chai W, Liu J, Jahn LA, Dale E, Fowler DE, Barrett EJ, et al. Salsalate attenuates free fatty acid-induced microvascular and metabolic insulin resistance in humans. Diabetes Care 2011; 34: 1634–1638.
- 7.
Watanabe S, Tagawa T, Yamakawa K, Shimabukuro M, Ueda S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol 2005; 25: 2376–2380.
- 8.
Azekoshi Y, Yasu T, Watanabe S, Tagawa T, Abe S, Yamakawa K, et al. Free fatty acid causes leukocyte activation and resultant endothelial dysfunction through enhanced angiotensin II production in mononuclear and polymorphonuclear cells. Hypertension 2010; 56: 136–142.
- 9.
Yasu T, Kobayashi M, Mutoh A, Yamakawa K, Momomura S, Ueda S. Dihydropyridine calcium channel blockers inhibit free fatty acid-induced endothelial and rheological dysfunction. Clin Sci 2013; 125: 247–255.
- 10.
Lipowsky HH. Microvascular rheology and hemodynamics. Microcirculation 2005; 12: 5–15.
- 11.
Fukuda S, Yasu T, Predescu DN, Schmid-Schönbein GW. Mechanisms for regulation of fluid shear stress response in circulating leukocytes. Circ Res 2000; 86: E13–E18.
- 12.
Fukuda S, Yasu T, Kobayashi, Ikeda N, Schmid-Shönbein GW. Contribution of fluid shear response in leukocytes to hemodynamic resistance in the spontaneously hypertensive rat. Circ Res 2004; 95: 100–108.
- 13.
Nishimura S, Manabe I, Nagasaki M, Seo K, Yamashita H, Hosoya Y, et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. J Clin Invest 2008; 118: 710–721.
- 14.
Yasu T, Ikeda N, Ishizuka N, Matsuda E, Kawakami M, Kuroki M, et al. Nicorandil and leukocyte activation. J Cardiovasc Pharmacol 2002; 40: 684–692.
- 15.
Rim SJ, Leong-Poi H, Lindner JR, Wei K, Fisher NG, Kaul S. Decrease in coronary blood flow reserve during hyperlipidemia is secondary to an increase in blood viscosity. Circulation 2001; 104: 2704–2709.
- 16.
Zambon A, Hashimoto SI, Brunzell JD. Analysis of techniques to obtain plasma for measurement of levels of free fatty acids. J Lipid Res 1993; 34: 1021–1028.
- 17.
Wada H, Yasu T, Kotsuka H, Hayakawa Y, Tsukamoto Y, Kobayashi N, et al. Evaluation of transmural myocardial perfusion by ultra-harmonic myocardial contrast echocardiography in reperfused acute myocardial infarction. Circ J 2005; 69: 1041–1046.
- 18.
Wei K, Jayaweera AR, Firoozan S, Linka A, Skyba DM, Kaul S. Quantification of myocardial blood flow with ultrasound-induced destruction of microbubbles administered as a constant venous infusion. Circulation 1998; 97: 473–483.
- 19.
Yoshioka T, Ageyama N, Shibata H, Yasu T, Misawa Y, Takeuchi K, et al. Repair of infarcted myocardium mediated by transplanted bone marrow-derived CD34+ stem cells in a nonhuman primate model. Stem Cells 2005; 23: 355–364.
- 20.
Kikuchi Y, Sato K, Ohki H, Kaneko T. Optically accessible microchannels formed in a single-crystal silicon substrate for studies of blood rheology. Microvasc Res 1992; 44: 226–240.
- 21.
Arao K, Yasu T, Umemoto T, Ikeda N, Ueda S, Kawakami M, et al. Effects of pitavastatin on fasting and postprandial endothelial function and blood rheology in patients with stable coronary artery disease. Circ J 2009; 73: 1523–1530.
- 22.
Ikeda N, Yasu T, Tsuboi K, Sugawara Y, Kubo N, Umemoto T, et al. Effects of submaximal exercise on blood rheology and sympathetic nerve activity. Circ J 2010; 74: 730–734.
- 23.
Cornelli U, Terranova R, Luca S, Cornelli M, Alberti A. Bioavailability and antioxidant activity of some food supplements in men and women using the D-Roms test as a marker of oxidative stress. J Nutr 2001; 131: 3208–3211.
- 24.
Vassalle C, Boni C, Cecco PD, Landi P. Elevated hydroperoxide levels as a prognostic predictor of mortality in a cohort of patients with cardiovascular disease. Int J Cardiol 2006; 110: 415–416.
- 25.
de Jongh RT, Serné EH, Ijzerman RG, de Vries G, Stehouwer CD. Free fatty acid levels modulate microvascular function: Relevance for obesity-associated insulin resistance, hypertension, and microangiopathy. Diabetes 2004; 53: 2873–2882.
- 26.
Liu J, Jahn LA, Fowler DE, Barrett EJ, Cao W, Liu Z. Free fatty acids induce insulin resistance in both cardiac and skeletal muscle microvasculature in humans. J Clin Endocrinol Metab 2011; 96: 438–446.
- 27.
Jayaweera AR, Wei K, Coggins M, Bin JP, Goodman C, Kaul S. Role of capillaries in determining CBF reserve: New insights using myocardial contrast echocardiography. Am J Physiol 1999; 277: H2363–H2372.
- 28.
Kaul S, Ito H. Microvasculature in acute myocardial ischemia: Part II evolving concepts in pathophysiology, diagnosis, and treatment. Circulation 2004; 109: 310–315.
- 29.
Tampakakis E, Tabit CE, Holbrook M, Linder EA, Berk BD, Frame AA, et al. Intravenous lipid infusion induces endoplasmic reticulum stress in endothelial cells and blood mononuclear cells of healthy adults. J Am Heart Assoc 2016; 5: e002574.
- 30.
Worthen GS, Schwab B III, Elson EL, Downey GP. Cellular mechanics of stimulated neutrophils: Stiffening of cells induces retention in pores in vitro and lung capillaries in vivo. Science 1989; 245: 183–185.
- 31.
Pleiner J, Schaller G, Mittermayer F, Bayerle-Eder M, Roden M, Wolzt M. FFA-induced endothelial dysfunction can be corrected by vitamin C. J Clin Endocrinol Metab 2002; 87: 2913–2917.
- 32.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000; 49: 1939–1945.
- 33.
Yu HY, Inoguchi T, Kakimoto M, Nakashima N, Imamura M, Hashimoto T, et al. Saturated non-esterified fatty acids stimulate de novo diacylglycerol synthesis and protein kinase C activity in cultured aortic smooth muscle cells. Diabetologia 2001; 44: 614–620.
- 34.
Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, et al. Vascular lipotoxicity: Endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology 2007; 148: 160–165.
- 35.
Uehara Y, Urata H, Sasaguri M, Ideishi M, Sakata N, Tashiro T, et al. Increased chymase activity in internal thoracic artery of patients with hypercholesterolemia. Hypertension 2000; 35: 55–60.
- 36.
Murakami K, Uehara Y, Abe S, Inoue Y, Ideishi M, Saku K, et al. Positive correlation between chymase-like angiotensin II-forming activity in mononuclear cells and serum cholesterol level. J Cardiol 2007; 50: 291–298.
- 37.
Kearney-Schwartz A, Virion JM, Stoltz JF, Drouin P, Zannad F. Haemorheological disturbances in hypertensive type 2 diabetic patients influence of antihypertensive therapy. Fundam Clin Pharmacol 2007; 21: 387–396.
- 38.
Arndt PG, Young SK, Poch KR, Nick JA, Falk S, Schrier RW, et al. Systemic inhibition of the angiotensin-converting enzyme limits lipopolysaccharide-induced lung neutrophil recruitment through both bradykinin and angiotensin II-regulated pathways. J Immunol 2006; 177: 7233–7241.
- 39.
Li H, Li H, Bao Y, Zhang X, Yu Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. Int J Cardiol 2011; 152: 218–224.
- 40.
Kamada T, McMillan DE, Sternlieb JJ, Bjork VO, Otsuji S. Erythrocyte crenation induced by free fatty acids in patients undergoing extracorporeal circulation. Lancet 1987; 2: 818–821.
- 41.
Reiss K, Cornelsen I, Husmann M, Gimpl G, Bhakdi S. Unsaturated fatty acids drive disintegrin and metalloproteinase (ADAM)-dependent cell adhesion, proliferation, and migration by modulating membrane fluidity. J Biol Chem 2011; 286: 26931–26942.


