Abstract
Background:
There is accumulating evidence that the AGEs-RAGE interaction plays an important role in accelerated atherosclerosis in diabetes. Our previous study showed that the AGEs-RAGE axis can reduce the cholesterol efflux of THP-1 macrophages through suppression of the expression of ABCG1 and that statins can inhibit the expression of RAGE. However, the role of statins in recovering the cholesterol efflux of macrophages reduced by AGEs has not been assessed.
Methods and Results:
ApoE−/−
mice and THP-1 macrophages were both treated by AGEs or AGEs combined with anti-RAGE antibody (only in THP-1 cells), ALT711 and atorvastatin separately. Cholesterol efflux of THP-1 macrophages and murine peritoneal macrophages was tested by fluorescence microplate technique. RT-PCR and western blot analysis were used to measure the expression of RAGE and molecules included in cholesterol efflux. After co-incubating with atorvastatin and AGEs, reduction in lipid accumulation in THP-1 macrophages and improvement of lesions complexity occurred compared with treating by AGEs only. Atorvastatin increased cholesterol efflux and ABCG1 expression of macrophages, which were reduced by AGEs, and decreased the expression of RAGE at the same time.
Conclusions:
This study demonstrated that atorvastatin can recover the deleterious ABCG1-mediated cholesterol efflux induced by AGEs in THP-1 macrophages and murine peritoneal macrophages by downregulating RAGE expression. It may contribute to the protective action of atorvastatin in diabetic subjects with atherosclerosis.
Atherosclerosis is a chronic inflammatory disease characterized by lipid and cholesterol accumulation within the walls of large and medium arteries.1
The accumulation of cholesterol-loaded macrophages (foam cell formation) in the intima of arteries is an early characteristic feature of atherosclerosis.2
The conversion of macrophages into foam cells is orchestrated by a disruption of the normal cholesterol homeostatic mechanism that controls the uptake, intracellular metabolism and efflux of cholesterol.3
Macrophage cholesterol efflux is a process that normally exports excess cholesterol from the cell for hepatic removal and is believed to be crucial for preventing atherogenesis and hence the development of most cardiovascular diseases.4
ATP-binding cassette transporter A1 (ABCA1) and G1 (ABCG1) play crucial roles in this process, in which accumulated cholesterol is removed by apolipoprotein A1 (ApoA1) and high-density lipoprotein (HDL), respectively, from the vessel wall to the liver for excretion.5
Thus, any impairment of the expression or functionality of the ABCG1 and ABCA1 transporters could have a significant effect on reverse cholesterol transport (RCT) and the development of atherosclerosis. ABCA1 and ABCG1 expressions are tightly regulated at both the transcriptional and posttranscriptional level. This regulation is mainly mediated by the nuclear oxysterol receptor, liver X receptor (LXR) and retinoid X receptor, which form heterodimers that are activated by oxysterols and retinoic acid, respectively.6
It is well established that people with diabetes mellitus (DM) have a greater risk of cardiovascular morbidity and mortality than their unaffected counterparts. More than 50% of DM-related deaths are associated with macrovascular complications, especially atherosclerosis.7
Advanced glycation endproducts (AGEs), a heterogeneous group of complex structures, form nonenzymatically when reducing sugars react with free amino groups on proteins, lipids, or nucleic acids.8
The formation and accumulation of AGEs progress under diabetic conditions.9
There is accumulating evidence that interaction between AGEs and the receptor for AGEs (RAGE) stimulates oxidative stress generation and subsequently evokes inflammatory and thrombogenic reactions, thereby being involved in accelerated atherosclerosis in DM.10,11
Some studies have also demonstrated that the AGEs-RAGE axis decreases ABCG1 or ABCA1 expression and reduces macrophage cholesterol efflux.12,13
Statins are competitive inhibitors for 3-hydroxy-3-methylglutaryl coenzyme A reductase, the rate-controlling enzyme in cholesterol synthesis. They are the most widely used cholesterol-lowering agents for treating the low-density lipoprotein-cholesterol (LDL-C) concentration and for the prevention of coronary artery disease. In clinical trials, statins have been proven to greatly reduce cardiovascular related morbidity and mortality.14,15
Besides their effects on lowering LDL-C, statins show pleiotropic effects, including improving endothelial function, enhancing the stability of atherosclerotic plaques, decreasing oxidative stress and inflammation, and inhibiting the thrombogenic response.16
Previous studies, including our own, report that statins can inhibit the expression of RAGE at both the RNA and protein level.17,18
Because our previous study demonstrated that the AGEs-RAGE axis can reduce the cholesterol efflux of THP-1 macrophages through suppression of the expression of ABCG1,19
we hypothesized that statins may recover the reduced macrophage cholesterol efflux elicited by AGEs through downregulating the expression of RAGE. However, there is also emerging evidence that statins reduce cholesterol efflux in macrophages through downregulating ABCA1 or ABCG1.20,21
The net sum of statins’ effects on cholesterol efflux in AGEs-pretreated macrophages is still unclear. Therefore, in this study, we examined whether atorvastatin, a widely used statin, could increase cholesterol efflux in AGEs-exposed THP-1 macrophages and murine peritoneal macrophages of ApoE−/−
mice and investigated the detailed mechanism.
Methods
Preparation of AGEs-BSA
Bovine serum albumin (BSA) and D-glucose were dissolved in phosphate-buffered saline (PBS: pH 7.2–7.4): the final concentrations of BSA and D-glucose were 5 g/L and 50 mmol/L, respectively. EDTA was added to a final concentration of 0.5 mmol/L to reduce oxidation. Penicillin (100 U/L) and streptomycin (100 μg/mL) were added to the reaction mixture to prevent bacterial contamination. The reaction mixture was filtered through 0.22-μm filter and then incubated at 37℃ for 12 weeks. At the end of the incubation period, the reaction mixture was dialyzed against sterilized PBS (pH 7.2–7.4) to remove the unconjugated glucose. The glucose level in the dialysate was <0.03 mmol/L. The reaction mixture was measured in a fluorospectrophotometer with an excitation wave of 370 nm, and the maximum absorption peak was measured at 440 nm to verify that the mixture was glycated BSA. Finally, the glycated BSA was freeze dried and stored at 4℃.
Mice and Diet
Male 8-week-old ApoE−/−
mice (on a C57BL/6 background) (Vital River Laboratory Animal Technology Co., Ltd, Beijing, China) were used in this study. At 8 weeks of age, mice were started on a non-irradiated high-fat diet (HFD: 16.6% lard, 1.3% cholesterol, 0.3% sodium cholate) for 10 weeks. Mice were housed in a specific pathogen-free facility on a 12 h light:dark cycle. Protocols were approved by the Institutional Animal Care and Use Committee of Tongji University.
Treatment in ApoE−/−
Mice
A total of 120 ApoE−/−
mice were randomly divided into 4 groups: (1) AGEs group received AGEs-BSA 30 mg/kg/day through intraperitoneal injection; (2) Statins+AGEs group was treated with AGEs-BSA 30 mg/kg/day through intraperitoneal injection and atorvastatin (Jia Lin Pharmaceutical Co. Ltd., Beijing, China) 10 mg/kg/day through intragastric administration; (3) ALT711+AGEs group received AGEs-BSA 30 mg/kg/day through intraperitoneal injection and ALT711, a type of cross-link breaker of AGEs (Ryan Chemical Co. Ltd., Suzhou, China) 1 mg/kg/day through intragastric administration; (4) control animals received the same amount of saline solution. At the end of week 10, all mice in the 4 groups were euthanased by intraperitoneal injection of pentobarbitone sodium (100 mg/kg body weight; Euthatal; Sigma-Aldrich, Castle Hill, NSW, Australia).
We chose the dose of atorvastatin mainly on the basis of our pilot study and the literature. It was higher than the human dose (1.1 mg/kg/day) recommended for treatment of hypercholesterolemia,22
but consistent with the pharmacokinetic data indicating a higher metabolic rate of the drug in rodents.23
Primary Macrophages Culture
Thioglycollate-induced peritoneal macrophages were obtained 4 days after an intraperitoneal injection of 1 mL 3% thioglycollate medium. Cells were harvested by flushing the peritoneal cavity with 10 mL ice-cold sterile PBS. Cells were seeded on tissue culture plates at a density of 1×106
cells/well in 24-well plates and 1×105
cells/well in NUNC Lab-Tek chamber slides (Thermo Scientific, Breda, The Netherlands). Cells were cultured in complete RPMI-1640 medium. After 3 h adhesion time, nonadherent cells were removed and the remaining peritoneal macrophages were analyzed. Immunofluorescence was used to identify macrophages.
Cell Culture and Treatment
The human monocyte cell line THP-1 was purchased from Scientific Research Institute (Shanghai, China) and cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 10 mmol/L HEPES (Sigma, USA), and 1% pen/strep solution at a density of 5×105
cells/mL in a 5% CO2
incubator. The cells were seeded in 6-well plates for 48 h in the presence of 100 ng/mL phorbol myristate acetate (PMA) (Sigma, USA), which allowed them to differentiate into adherent macrophages. The culture medium was then changed to RPMI1640 medium containing 0.1% FBS for 6 h of cell starvation. The macrophages were pretreated with BSA (600 μg/mL) as the control group or with AGEs (600 μg/mL) for 2 h, then stimulated with oxidized LDL (oxLDL: 100 μg/mL; Jingke Chemistry, China) for 24 h at 4℃ in a 5% CO2
incubator, and then collected for detection. To observe the effect of atorvastatin, 4 groups of cells were treated with atorvastatin (48 mg/L), anti-RAGE antibody (10 μg/mL; Abcam, USA) and ALT711 (20 μg/mL), and anti-RAGE antibody plus atorvastatin, respectively, for 2 h before adding AGEs.
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen, CA, USA). For reverse transcription (RT), 2 μg of the total RNA was converted to first-strand complementary DNA in 20-μL reactions using a RT kit (Thermo, USA). Quantitative real-time PCR analysis was performed (StepOne, Applied Biosystems) using SYBR Green (Takara, Japan). The thermal cycling program was 10 min at 95℃ for enzyme activation and 40 cycles of denaturation for 15 s at 95℃, 5 min at 60℃ for annealing and extension. The comparative cycle threshold method was used to determine relative mRNA expressions of genes as normalized by the GAPDH housekeeping gene. Gene expression was determined by the 2−∆∆Ct
method. All primers used are shown in
Table 1.
Table 1.
PCR Primer Sequence
| Gene name |
Primer sequence 5’–3’ |
| Human |
| RAGE |
GAAACTGAACACAGGCCGGA |
| ABCG1 |
TGTCTGATGGCCGCTTTCT |
| ABCA1 |
AGGGAGAG CACAGGCTTTGAC |
| LXRα |
GATTACAACGGTGATGGCGG |
| GAPDH |
TGACGGGATCTCGCTCCTGGAAGAT |
| Murine |
| RAGE |
TCTCCGCTTCCTCTGACTGA |
| GAPDH |
AGCAGTCCCGTACACTGGCAAAC |
Cells described before were lysed with RIPA lysis buffer (50 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing a protease inhibitor cocktail (Sigma, USA), and the protein concentrations of lysates were determined by the BCA assay kit (Beyotime, China). Proteins (20 μg of extracts) were electrophoresed by 12% SDS-PAGE, and transferred to a nitrocellulose membrane. The membranes were blocked overnight at 4℃ in 5% skim milk, and then incubated with primary antibodies against RAGE, ABCA1, ABCG1, and LXRα (1:1,000) (Abcam, USA) for 2 h at room temperature. The membranes were washed with tris buffered saline added with tween (TBS-T), and incubated with a horseradish peroxidase conjugated secondary anti-mouse or anti-rabbit IgG antibody (1:5,000, Bioss, China) for 1 h at 37℃. Afterwards blots were developed using ECL Plus. The signal intensities of specific bands were detected with a Clinx ChemiScope imaging-system (Qinxiang, China) and quantified using TotalLab TL100-Quick Start analysis software.
Oil Red O Staining in THP-1 Macrophages
After the aforementioned treatment, the THP-1 differentiated macrophages were collected, washed twice with ice-cold PBS, and then fixed with 10% neutral-buffered formalin for 15 min. The cells were washed again with 60% isopropyl alcohol for 1 min and then stained with oil red O (Sigma, USA) for 15 min. The oil red O was discarded, and excess dye was washed away with 60% isopropyl alcohol. After washing with distilled water, the specimens of each group were placed under an inverted microscope to observe oil red staining. Cellular lipid accumulation was quantified by the optical density (OD) at 518 nm of oil red O extracted from stained cells using isopropanol. The results were normalized as OD value/number of cells.
Cholesterol Efflux Assay
The treated THP-1 differentiated macrophages and murine peritoneal macrophages were plated in 12-well plates and loaded with 40 μg/mL oxLDL labeled with 1,1’-dioctadecyl-3,3,3’,3’-tetramethyl-indocarbocyanine perchlorate (DiI-oxLDL) for 48 h at room temperature. The medium was then removed and cells were washed twice with PBS. The culture medium was then changed to RPMI1640 medium containing 0.1% BSA supplemented with ApoAI (10 μg/mL; Novoprotein, China) or HDL (100 μg/mL; Jingke Chemistry, China) at 37℃ for 24 h. At the end of this incubation period, the supernatant was collected and centrifuged at 12,000 g for 10 min to remove debris. Cells were lysed with 0.5 mL of 0.1 N NaOH. The fluorescence of both the supernatant and cellular lipid was measured on a fluorescence microplate (Thermo Labsystems, USA). Fluorescence intensity was determined and expressed as a percentage of the total cell DiI-oxLDL content (effluxed DiI-oxLDL+cell-associated DiI-oxLDL).
Atherosclerosis Study
ApoE−/−
mice were fed HFD for 10 weeks and then killed humanely by an intraperitoneal injection of pentobarbitone sodium (100 mg/kg body weight). Aortas were perfused with PBS, isolated and fixed in neutral phosphate-buffered formalin. The aortic arch and descending aorta were stained with oil red O. Aortas were pinned on silicon dishes and the oil red O-positive areas were quantified using Image J software and expressed as the percentage of the total aortic area. The aortic root area was cross-sectioned in 5-μm sections, which were stained with hematoxylin and eosin and the average of 6 evenly distributed sections for each animal was used to determine lesion size and complexity. Lesion size was quantified by morphometric analysis using Image-Pro Plus software (Media Cybernetics). The typing of lesions was done according to the typing for humans proposed by the American Heart Association24
and adapted to categorize murine lesions.25
In this study, we discerned sections showing macrophage foam cell-rich lesions (types I–II), complex lesions with fibrous caps (type III), and advanced lesions with foam cells in the media and presence of fibrosis, cholesterol clefts, mineralization and/or necrosis (types IV–V).
Measurements of Serum Fast Glucose, Lipids and AGEs of ApoE−/− Mice
Serum glucose levels were measured by the glucose oxidase method (Sigma, MO, USA). Total cholesterol (TC), LDL-C, high-density lipoprotein-cholesterol (HDL-C) and the L/H ratio were measured using a kit from Sigma Diagnostics. Serum AGEs were measured using a competitive ELISA Kit with an immunopurified anti-AGEs antibody (Jiancheng Bioengineering Institute, Nanjing, China). The procedure was performed as per the manufacturer’s instructions.
Statistical Analysis
Data are expressed as mean±SD. Results were analyzed by χ2-test or one-way ANOVA followed by Fisher’s least significant difference (LSD) or Dunnett’s T3 procedure using SPSS 21.0. P<0.05 was considered statistically significant. All experiments were performed at least 3 times.
Results
Metabolic Parameters
At the end of the 10th week, serum fasting blood glucose, TC, LDL-C, and HDL-C levels and L/H ratio were not different among the 4 groups of ApoE−/−
mice (Table 2).
Table 2.
Metabolic Parameters of ApoE
−/− Mice in Different Groups
| |
Fasting glucose |
TC |
LDL |
HDL |
L/H ratio |
| Control |
7.30±2.05 |
22.19±1.10 |
20.07±1.17 |
3.93±0.29 |
5.11±0.35 |
| AGEs |
7.17±1.06 |
27.53±1.72 |
25.77±7.61 |
4.85±0.84 |
5.23±0.83 |
| AGEs+ALT711 |
6.99±1.60 |
26.23±0.93 |
24.23±3.12 |
4.40±0.69 |
5.56±0.73 |
| AGEs+atorvastatin |
6.70±0.63 |
26.66±2.68 |
26.41±2.12 |
4.42±0.62 |
5.98±0.85 |
Each value represents the mean±SD. AGEs, advanced glycation endproducts.
In THP-1 differentiated macrophages, RAGE expression was substantially upregulated at both the mRNA and protein level after incubation with AGEs compared with the control group. After pretreating with anti-RAGE antibody, ALT711 and atorvastatin, RAGE expression was suppressed at both the mRNA and protein level compared with the AGEs group, and there were no significant differences among the 3 groups with pretreatment at the mRNA level; however, at the protein level RAGE expression in the anti-RAGE antibody group was lower than in the other 2 groups. When pretreated with atorvastatin combined with anti-RAGE antibody, the expression of RAGE was even lower than when pretreated with anti-RAGE antibody only (Figure 1A,B). Similar to our in vitro data, an increase in RAGE expression in peritoneal macrophages of ApoE−/−
mice could be observed after AGEs treatment as compared with the control group, and atorvastatin reversed the increase (Figure 1C,D).

We observed that treatment of THP-1 macrophages with AGEs promoted foam cell formation, because their neutral lipid content was significantly increased compared with untreated macrophages. However, this trend was inhibited by atorvastatin pretreatment to the same extent as with anti-RAGE antibody and ALT711 (Figure 2).
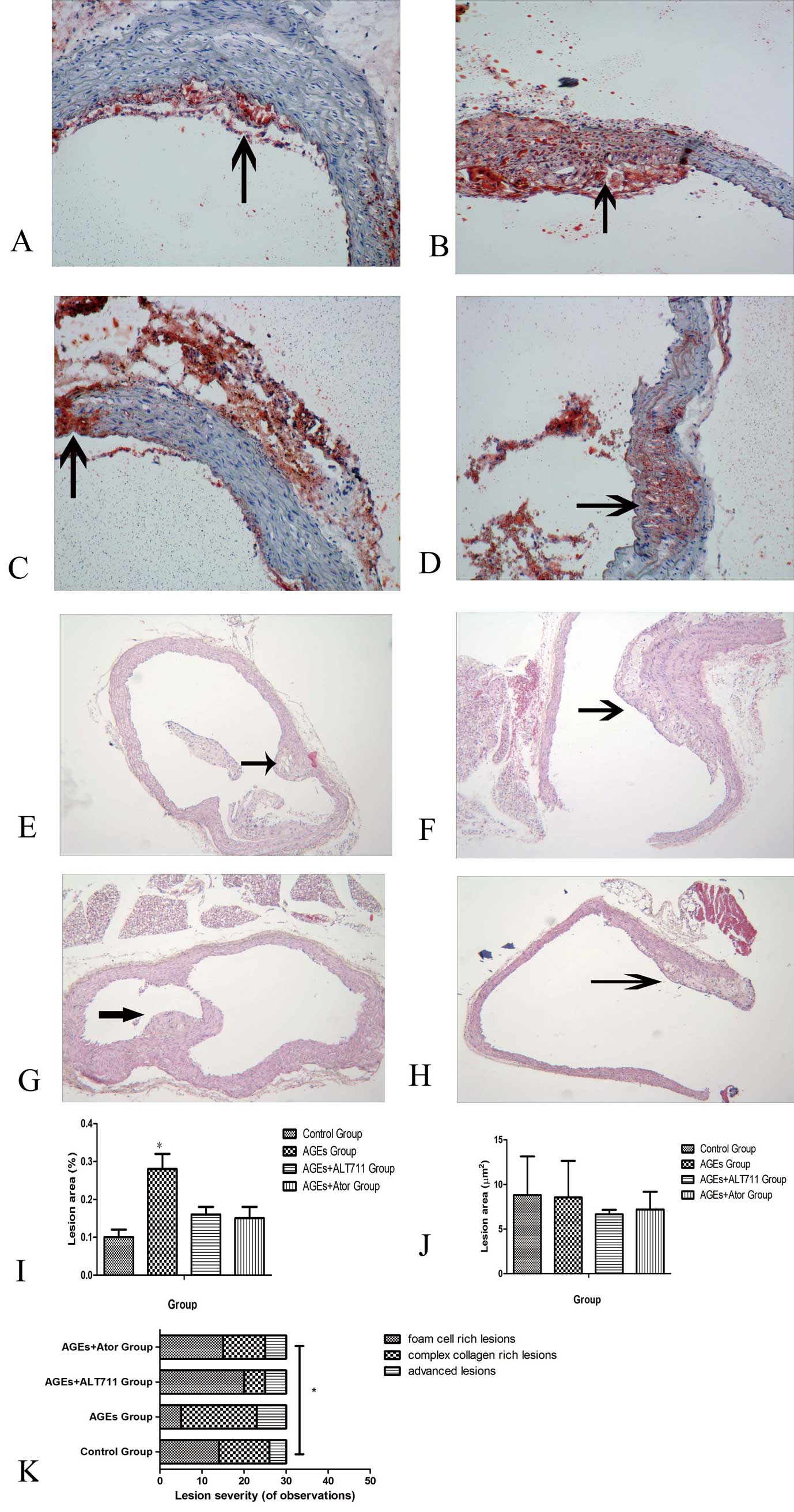
Effect of Atorvastatin on AGEs-Induced Atherosclerosis in Aortas of ApoE−/− Mice
Atherosclerotic lesion area as evaluated by oil red O staining of the aortic branch area was significantly reduced in atorvastatin-treated mice compared with only AGEs-treated mice (Figure 3A–D,I). Although there was no difference in the overall lesion area in the aortic root (Figure 3J), atherosclerosis was less advanced as judged by reduced lesion complexity in atorvastatin-treated mice than only AGEs-treated mice (Figure 3K). Thus, atorvastatin conferred protection from atherosclerosis induced by AGEs.
Effects of Atorvastatin on HDL-Mediated Cholesterol Efflux in Macrophages Treated by AGEs
We examined whether the increase in lipids in THP-1 differentiated macrophages upon AGEs inducement could be the result of decreased cholesterol efflux. In macrophages, cholesterol is transferred to ApoA1 or to HDL particles for RCT.5
As shown in
Figure 4A,B, AGEs inducement resulted in a diminished HDL-mediated cholesterol efflux in THP-1 macrophages, whereas ApoA-1-mediated efflux remained unaffected. Macrophage cholesterol efflux to ApoA1 is mediated via ABCA1, whereas efflux to HDL is mediated via ABCG1. Correspondingly, we also found a significant decrease in ABCG1 but not ABCA1 gene expression in lipid-loaded macrophages upon AGEs inducement (Figure 4C,D). Moreover, ABCG1 protein levels were also found to be downregulated, as shown by western blot analysis (Figure 4G). After treatment with atorvastatin as well as anti-RAGE antibody and ALT711, HDL-mediated cholesterol efflux was increased in parallel with the upregulation of ABCG1 expression compared with the AGEs-induced group, but the recovering effect of atorvastatin was weaker than anti-RAGE antibody and ALT711. When the cells were exposed to both atorvastatin and anti-RAGE antibody, ABCG1 expression did not increase anymore compared with exposure to anti-RAGE antibody only (Figure 4C–D,F–G). Next we detected the expression of the LXR, the classical regulator of ABCG1, and found that LXR expression was inconsistent with that of ABCG1 at both the RNA and protein level (Figure 4E,H).
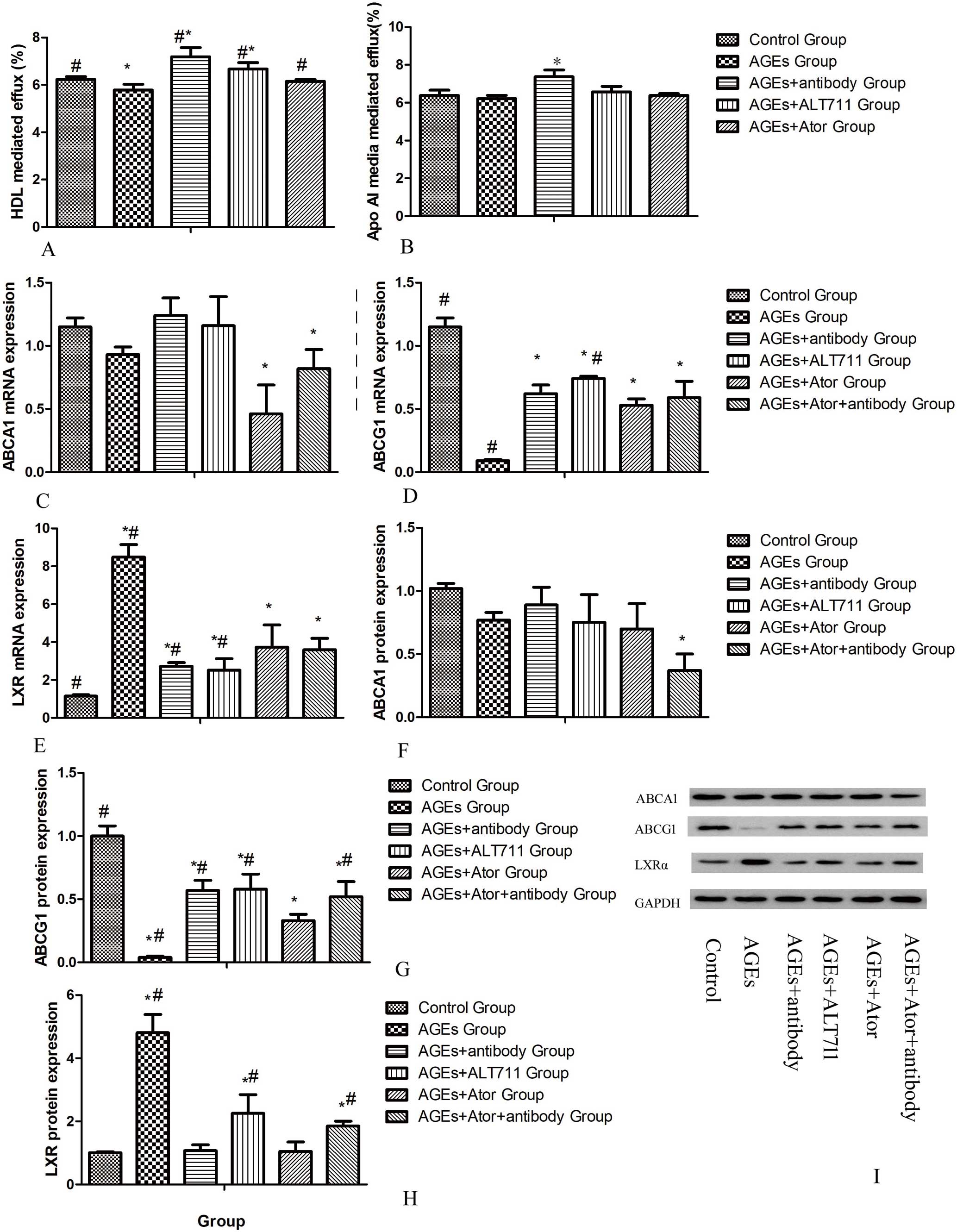
To confirm our results in vivo, ApoE−/−
mice were put on a high cholesterol diet and treated respectively by AGEs, AGEs plus atorvastatin or ALT711 for a period of 10 weeks, and peritoneal macrophages were then obtained. Similar to our in vitro data, a decrease in both HDL-mediated cholesterol efflux and in ABCG1 protein expression was observed in the AGEs-treated group as compared with the control group (Figure 5B,D). As expected, atorvastatin as well as ALT711 normalized the changes induced by AGEs, both in HDL-mediated cholesterol efflux and ABCG1 protein expression (Figure 5B,D).
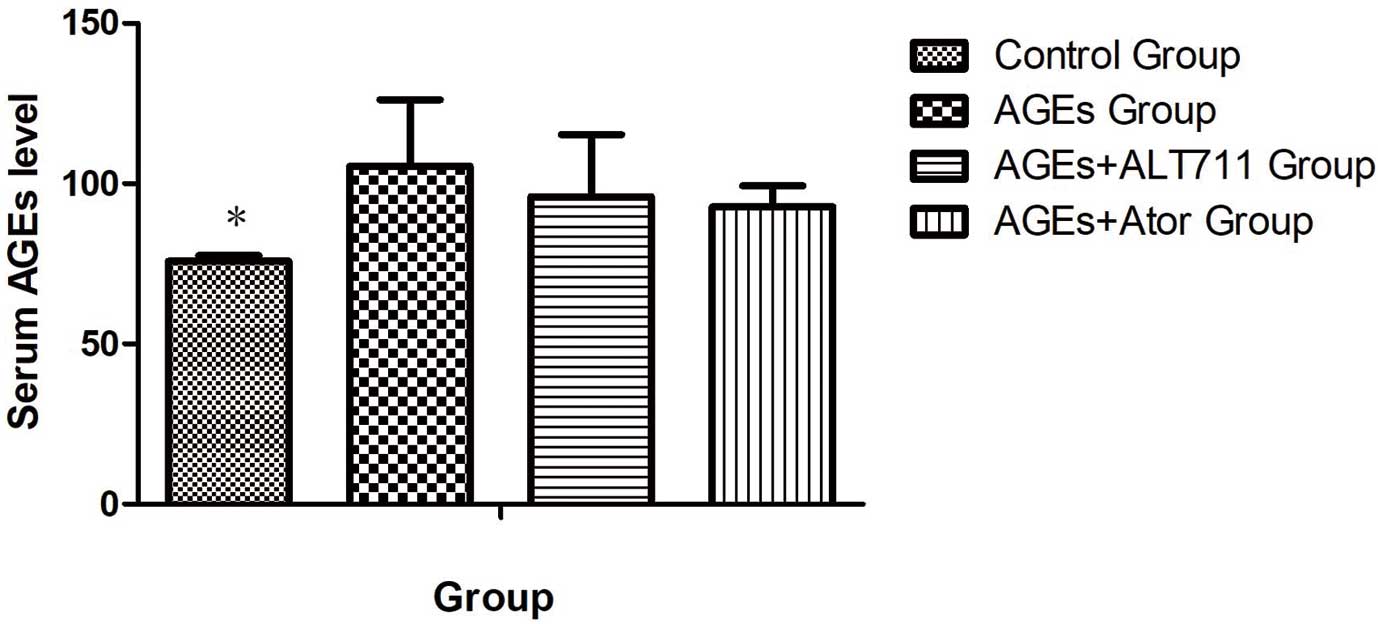
Effect of Atorvastatin Treatment on the Serum AGEs Level in ApoE−/−
Mice
Serum AGEs were significantly higher in the AGEs-treated group compared with the control group. Administration of atorvastatin to AGEs-treated ApoE−/−
mice moderately reduced the serum AGEs level without statistical significance (Figure 6).
Discussion
In the present study, we confirmed that AGEs-RAGE interaction reduced macrophage cholesterol efflux mainly through downregulating ABCG1 expression in both our in vitro and in vivo studies. Treatment with atorvastatin increased macrophage cholesterol efflux, which was reduced by AGEs through increasing ABCG1 expression both in vitro and in vivo and improved atherogenesis in ApoE−/−
mice.
The ABCG1 transporter mediates cholesterol efflux to HDL particles, which contributes to 30% of total cholesterol efflux from macrophages and thus plays a key role in macrophage RCT and lipid homeostasis.26
In a previous study, we addressed that AGEs reduced cholesterol efflux through HDL but not ApoAI in THP-1 macrophages through binding with RAGE. Reports from our group showed that AGEs mainly reduced the expression of ABCG1 but not ABCA1 in an LXRα-independent manner in THP-1 macrophages, supporting the notion that ABCG1 may be especially important in diabetic atherosclerosis.19
To further understand the in vivo role of the AGEs-RAGE axis, in the present study we treated ApoE−/−
mice with an injection of AGEs-BSA and noted that serum AGEs levels were obviously higher than in the control group after injection, and we found cholesterol efflux mediated by ABCG1 was obviously reduced in peritoneal macrophages of ApoE−/−
mice, which was also not regulated by LXRα, and more lipid accumulation was observed in the aortas of ApoE−/−
mice after AGEs-BSA injection. The results of our present study confirmed the adverse effects of the AGEs-RAGE axis in reducing macrophage cholesterol efflux through special signaling pathways without dependence on LXRα. Accumulation of AGE proteins is thus an atherosclerotic complication in diabetic patients by reducing macrophage cholesterol efflux.
An increasing body of evidence from in vitro studies has demonstrated downregulatory effects of statins on ABCA1 and ABCG1 expression and functionality in macrophages,21,27
which seems a deleterious effect of statins on RCT in contrast to their beneficial lipid-lowering effects. However, our results are in disagreement with the results of previous reports. Our study observed recovery of ABCG1 expression and macrophage cholesterol efflux mediated by HDL after atorvastatin treatment both in vitro and in vivo when pretreating THP-1 macrophages and ApoE−/−
mice with AGEs-BSA. The disparities can be mainly attributed to the AGEs-BSA exposure of macrophages in our study. AGEs are often increased in aging, but mainly in poorly controlled DM and are involved in accelerated atherosclerosis in DM.10,28,29
Our study indicated that statins may be more protective in diabetic patients than in patients with normal glucose metabolism. Both the Heart Protection Study (HPS) and Collaborative Atorvastatin Diabetes Study (CARDS) confirmed that statin therapy can reduce cardiovascular events significantly in diabetic patients, which is mainly related to LDL-C reduction.30,31
Our results showed a special beneficial effect of atorvastatin in diabetic patients that may normalize the macrophage RCT inhibited by AGEs. Besides the increase in ABCG1 expression and HDL-mediated macrophage cholesterol efflux, our study also showed the reduction of lipid accumulation in THP-1 macrophages and the aorta after atorvastatin treatment. Although there was no difference in overall lesion area in the proximal aorta, atherosclerosis was less advanced as judged by reduced lesion complexity in the ApoE−/−
mice in the atorvastatin-treated group. Thus, atorvastatin therapy conferred protection from atherosclerosis induced by AGEs, which may be attributed to the improvement of macrophage cholesterol efflux. In order to understand the extent of the effect of atorvastatin well, ALT711, a type of cross-link breaker of AGEs, and anti-RAGE antibody were used as the control. Our findings showed that atorvastatin’s effect was a little weaker than that of ALT711 or anti-RAGE antibody in both ABCG1 reduction and HDL-mediated macrophage cholesterol efflux. Because there are no drugs for breaking AGEs or blocking AGEs-RAGE interaction that can be used clinically, atorvastatin as a widely used high-intensity statin may be a choice for improving macrophage RCT in diabetic patients.
Necessarily, the mechanism of atorvastatin on ABCG1 expression needs to be determined. Despite the described positive correlation between the expression of LXRα and the studied RCT genes,32
no correlation was observed between LXRα and ABCG1 in our study. Thus, the upregulatory effects of atorvastatin on ABCG1 expression were unlikely because of any change in LXRα. This is in accordance with our results showing that atorvastatin had no effect on the upregulation of LXRα expression. In light of these findings, there are 2 possible pathways through which atorvastatin can play its role in ABCG1 regulation. One is that atorvastatin has a direct effect on ABCG1 upregulation; however, that is in disagreement with most of the previous studies.20,27
In order to observe a direct effect of atorvastatin on ABCG1 regulation, we used anti-RAGE antibody to block AGEs-RAGE interaction in THP-1 macrophages before treating them with atorvastatin. Compared with the group treated with anti-RAGE antibody alone, ABCG1 expression was not further increased in the group treated with anti-RAGE antibody plus atorvastatin. This result indicated that atorvastatin may not have a direct effect on increasing ABCG1 expression when AGEs-RAGE interaction is blocked. The other pathway is that atorvastatin increases ABCG1 expression through downregulating the expression of RAGE in macrophages. An inhibiting effect of atorvastatin on RAGE expression was observed in our present study, and in our previous study we also reported that atorvastatin downregulated RAGE, possibly by modifying the RAGE gene through an epigenetic mechanism.23
In the present study, ABCG1 expression in macrophages after treatment with atorvastatin did not show the same trend as ABCA1. ABCA1 expression was decreased in AGEs-induced macrophages after treatment with atorvastatin, which is consistent with most other studies. However, ABCG1 expression increased under the same conditions to a weaker extent than in the anti-RAGE antibody or in ALT711-treated groups. It is tempting to speculate that atorvastatin may regulate ABCG1 indirectly. This point is strengthened by our previous findings that the AGEs-RAGE axis could inhibit RCT mainly through decreasing ABCG1 but not ABCA1 expression. We further detected the serum level of AGEs in ApoE−/−
mice, and found that atorvastatin did not significantly reduce the serum level of AGEs induced by AGEs-BSA injection. That may also imply atorvastatin directly downregulates RAGE expression without changing the serum level of AGEs. On this point, the relevance of the effects of atorvastatin on ABCG1 functionality in the context of RAGE−/−
animal models may need further investigation.
In summary, atorvastatin can recover the deleterious ABCG1-mediated cholesterol efflux induced by AGEs in THP-1 macrophages and murine peritoneal macrophages. The upregulatory effects of atorvastatin on ABCG1 functionality are likely caused by the downregulation of RAGE expression. Therefore, the present study provides an additional beneficial aspect of atorvastatin in DM. Atorvastatin may prevent the development and progression of atherosclerosis in DM by not only reducing serum cholesterol level, but also by improving cholesterol efflux from foam cells of the arterial wall via blocking the harmful effects of the AGEs-RAGE axis on macrophages. Whether this is cause for concern clinically remains to be established.
Acknowledgments
This work was partly supported by the Program of Shanghai Municipal Commission of Health and Family Planning for Medical Research and Development under Grant number (201640387), the Fundamental Research Funds for the Central Universities under Grant number (1507219062) and grants from National Natural Science Foundation of China (81300699).
References
- 1.
Lusis AJ. Atherosclerosis. Nature 2000; 407: 233–241.
- 2.
Pamukcu B, Lip GY, Devitt A, Griffiths H, Shantsila E. The role of monocytes in atherosclerotic coronary artery disease. Ann Med 2010; 42: 394–403.
- 3.
Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron 2006; 37: 208–222.
- 4.
Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med 2002; 8: 1235–1242.
- 5.
Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 2010; 30: 139–143.
- 6.
Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci USA 2000; 97: 12097–12102.
- 7.
Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: A Statement for Healthcare Professionals from the American Heart Association. Circulation 1999; 100: 1134–1146.
- 8.
Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I: The AGE concept. Cardiovasc Res 1998; 37: 586–600.
- 9.
Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med 1988; 318: 1315–1321.
- 10.
Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res 2010; 106: 842–853.
- 11.
Yamagishi S, Matsui T. Smooth muscle cell pathophysiology and advanced glycation end products (AGEs). Curr Drug Targets 2010; 11: 875–881.
- 12.
Passarelli M, Tang C, McDonald TO, O’Brien KD, Gerrity RG, Heinecke JW, et al. Advanced glycation end product precursors impair ABCA1-dependent cholesterol removal from cells. Diabetes 2005; 54: 2198–2205.
- 13.
Isoda K, Folco EJ, Shimizu K, Libby P. AGE-BSA decreases ABCG1 expression and reduces macrophage cholesterol efflux to HDL. Atherosclerosis 2007; 192: 298–304.
- 14.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366: 1267–1278.
- 15.
LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: A meta-analysis of randomized controlled trials. JAMA 1999; 282: 2340–2346.
- 16.
Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017; 120: 229–243.
- 17.
Xu L, Zang P, Feng B, Qian Q. Atorvastatin inhibits the expression of RAGE induced by advanced glycation end products on aortas in healthy Sprague-Dawley rats. Diabetol Metab Syndr 2014; 6: 102.
- 18.
Prasad K, Tiwari S. Therapeutic interventions for advanced glycation-end products and its receptor-mediated cardiovascular disease. Curr Pharm Des 2017; 23: 937–943.
- 19.
Xu L, Wang YR, Li PC, Feng B. Advanced glycation end products increase lipids accumulation in macrophages through upregulation of receptor of advanced glycation end products: Increasing uptake, esterification and decreasing efflux of cholesterol. Lipids Health Dis 2016; 15: 161.
- 20.
Wong J, Quinn CM, Gelissen IC, Jessup W, Brown AJ. The effect of statins on ABCA1 and ABCG1 expression in human macrophages is influenced by cellular cholesterol levels and extent of differentiation. Atherosclerosis 2008; 196: 180–189.
- 21.
Genvigir FD, Hirata MH, Hirata RD. ABCA1 expression and statins: Inhibitory effect in peripheral blood mononuclear cells. Pharmacogenomics 2009; 10: 997–1005.
- 22.
Bernini F, Poli A, Paoletti R. Safety of HMG-CoA reductase inhibitors: Focus on atorvastatin. Cardiovasc Drugs Ther 2001; 15: 211–218.
- 23.
Dostal LA, Whitfield LR, Anderson JA. Fertility and general reproduction studies in rats with the HMG-CoA reductase inhibitor, atorvastatin. Fundam Appl Toxicol 1996; 32: 285–292.
- 24.
Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995; 92: 1355–1374.
- 25.
Gijbels MJ, van der Cammen M, van der Laan LJ, Emeis JJ, Havekes LM, Hofker MH, et al. Progression and regression of atherosclerosis in APOE3-Leiden transgenic mice: An immunohistochemical study. Atherosclerosis 1999; 143: 15–25.
- 26.
Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, et al. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res 2007; 48: 2453–2462.
- 27.
Wang W, Song W, Wang Y, Chen L, Yan X. HMG-CoA reductase inhibitors, simvastatin and atorvastatin, downregulate ABCG1-mediated cholesterol efflux in human macrophages. J Cardiovasc Pharmacol 2013; 62: 90–98.
- 28.
Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006; 114: 597–605.
- 29.
Jandeleit-Dahm K, Cooper ME. The role of AGEs in cardiovascular disease. Curr Pharm Des 2008; 14: 979–986.
- 30.
Collins R, Armitage J, Parish S, Sleigh P, Peto R; Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: A randomised placebo-controlled trial. Lancet 2003; 361: 2005–2016.
- 31.
Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004; 364: 685–696.
- 32.
Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta 2013; 424: 245–252.