Abstract
Background:
Ischemic preconditioning (IPC) is an effective procedure to protect against ischemia/reperfusion (I/R) injury. Hypoxia-inducible factor-1α (Hif-1α) is a key molecule in IPC, and roxadustat (RXD), a first-in-class prolyl hydroxylase domain-containing protein inhibitor, has been recently developed to treat anemia in patients with chronic kidney disease. Thus, we investigated whether RXD pretreatment protects against I/R injury.
Methods and Results:
RXD pretreatment markedly reduced the infarct size and suppressed plasma creatinine kinase activity in a murine I/R model. Analysis of oxygen metabolism showed that RXD could produce ischemic tolerance by shifting metabolism from aerobic to anaerobic respiration.
Conclusions:
RXD pretreatment may be a novel strategy against I/R injury.
Early reperfusion is the most effective therapy to reduce infarct size and preserve cardiac function after myocardial infarction, and revascularization techniques such as percutaneous coronary intervention (PCI) and coronary artery bypass grafting (CABG) are well established. Nevertheless, ischemic heart disease (IHD) remains a leading cause of heart failure.1
New strategies to manage IHD are required for further improvement of cardiovascular disease outcomes.
Editorial p 891
Ischemic preconditioning (IPC) is an effective procedure to protect against ischemia/reperfusion (I/R) injury, and remote IPC (RIPC), developed from IPC, is a promising strategy to prevent I/R injury.2
However, 2 large randomized trials, the ERICCA trial and the RIPHeart study, failed to find clinical benefits from RIPC during elective CABG.3,4
Moreover, Hausenloy et al recently reported that RIPC did not improve clinical outcomes at 12 months in patients with STEMI undergoing PCI.5
Current evidence indicates that another approach is necessary for clinical application of IPC to I/R injury.
To pharmacologically reproduce IPC, several potential meditators of IPC, including adenosine, protein kinase C, opioid receptor, and reactive oxygen species, have been identified.6
Hypoxia-inducible factor-1α (Hif-1α), induced by the inhibition of prolyl hydroxylase domain-containing proteins (PHDs) under hypoxia, plays a key role in IPC.7
Under well-oxygenated conditions, PHDs hydroxylate Hif-1α and hydroxylated Hif-1α is subsequently bound by von Hippel-Lindau (VHL), which recruits ubiquitin ligase, resulting in ubiquitination and proteasome degradation.8
Conversely, under hypoxic conditions, Hif-1α hydroxylation ceases, as does ubiquitination and degradation.9
As a result, Hif-1α accumulates intracellularly and functions as a transcriptional and non-transcriptional factor.9,10
Minamishima and Kaelin reported that PHD inhibition upregulates erythropoietin and promotes red blood cell production in the liver,11
which led to the development of a first-in-class PHD inhibitor, roxadustat (RXD), to treat anemia in patients with chronic kidney disease.12
However, the efficacy of RXD in myocardial I/R injury remains to be fully elucidated. Thus, we investigated whether RXD pretreatment protects against myocardial I/R injury in mice.
Methods
Murine I/R Model
All procedures involving animals and animal care protocols were approved by the Committee on Ethics of Animal Experiments of the Kyushu University Graduate School of Medical and Pharmaceutical Sciences (A19-028). In the murine model of I/R, 9–12-week-old male C57BL/6J mice (Kyudo Co. Ltd., Saga, Japan) were anesthetized with isoflurane, the intercostal space was opened under mechanical ventilation, and ischemia was induced by ligation of the left anterior descending coronary artery (LAD) for 45 min, followed by reperfusion. RXD (HY-13426, MedChemExpress, NJ, USA), dissolved in DMSO, was subcutaneously administered. Plasma creatinine kinase (CK) activity was measured using the Colorimetric Assay Kit (BioVision, K777, CA, USA). Tissue sections of the left ventricle (LV) were stained with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining (Takara, Shiga, Japan) in accordance with manufacturer’s instructions.
Western Blotting
Western blotting was performed as previously described.13
Primary antibodies against the following proteins were used: Hif-1α (#36169, Cell Signaling Technology, MA, USA), pyruvate kinase isozyme M2 (PKM2; #32532, Cell Signaling Technology), protein regulated in development and DNA damage response 1 (REDD1; 10638-1-AP, Proteintech, IL, USA) and lactate dehydrogenase (LDH; #9579, Cell Signaling Technology). An antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; sc-32233, Santa Cruz Biotechnology, CA, USA) was used as a control.
Quantitative Reverse Transcription PCR (qRT-PCR)
qRT-PCR was performed as previously described.13
The forward and reverse primer sequences, respectively, were (5′–3′):
Dtit4, TGGACAGCAGCAACAGTGG and TGCATCAGGTTGGCACACAG;
Ndufa4l2, CTGGGACAAGATGGCAGGAAC and GGGCAAGTCGCAGCAAGTAGA; and
Rps18, TTCTGGCCAACGGTCTAGACAAC and CCAGTGGTCTTGGTGTGCTGA.
Cell Culture and XFp Analysis
Cultured cardiomyocytes were prepared as described previously.13
Lipofectamine RNAiMAX (Thermo Scientific, MA, USA) was used for transfection of siRNA in accordance with the manufacturer’s instructions. siRNAs against Hif-1α (RS0035615 and RS0035613, used at 1 nmol/L as final concentration) were purchased from Takara Bio Inc (Shiga, Japan). Oxygen consumption rate ([OCR] representing aerobic respiration) and extracellular acidification rate ([ECAR] representing anaerobic respiration) in cultured cardiomyocytes were measured under RXD treatment using XFp analysis (Agilent Technologies, CA, USA), as previously described.14
Cell Survival Assay After Hypoxia/Reoxygenation (H/R)
Hypoxia in cultured cardiomyocytes was induced by replacing standard medium with conditioned medium that included 25 μmol/L RXD in a hypoxic chamber (APM-50D, ASTEC, Fukuoka, Japan; 1% O2, 5% CO2, and N2) overnight. Subsequently, a 96-well plate was promptly transferred into the hypoxic chamber (1% O2) and incubated for 4 h. After hypoxia, the 96-well plate was incubated in normoxia for 1 h as a reoxygenation condition. Pretreatment with RXD was started 4 h before induction of hypoxia. Cell survival was measured using a Cell Counting Kit-F (Dojindo, Kumamoto, Japan) in accordance with the manufacturer’s instructions.
Statistical Analysis
Data are shown as mean±standard error of the mean (SEM); P-value <0.05 was considered significant by Student’s t-test, Dunnett’s test, and one-way ANOVA with a post-hoc test (Tukey test). JMP14 software (SAS Institute, NC, USA) was used for the statistical analysis.
Results
Cardioprotection of RXD Pretreatment Against I/R Injury
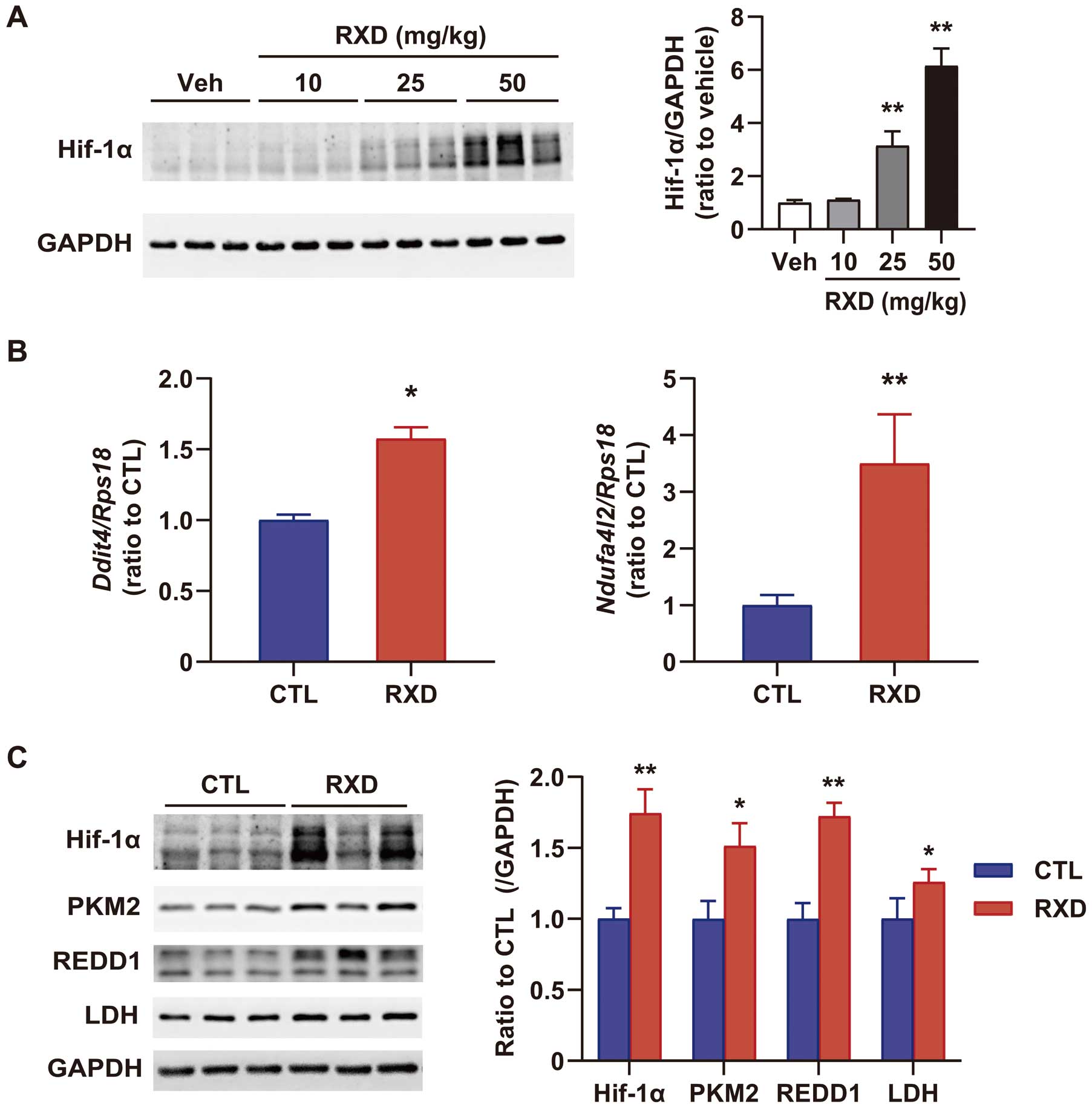
The expression of Hif-1α in the myocardium increased in a dose-dependent manner 3 h after RXD treatment (10 mg/kg, 25 mg/kg, and 50 mg/kg) (Figure 1A). In this study, RXD was used at 25 mg/kg, which is the standard dose to increase hemoglobin concentration in mice (unpublished data, Interview Form of Evrenzo®, https://amn.astellas.jp/jp/di/list/evz/if_evz.pdf [in Japanese]). A single administration of RXD (25 mg/kg, s.c.) in mice upregulated hypoxia-responsive genes, such as
Ddit4
and
Ndufa4l2
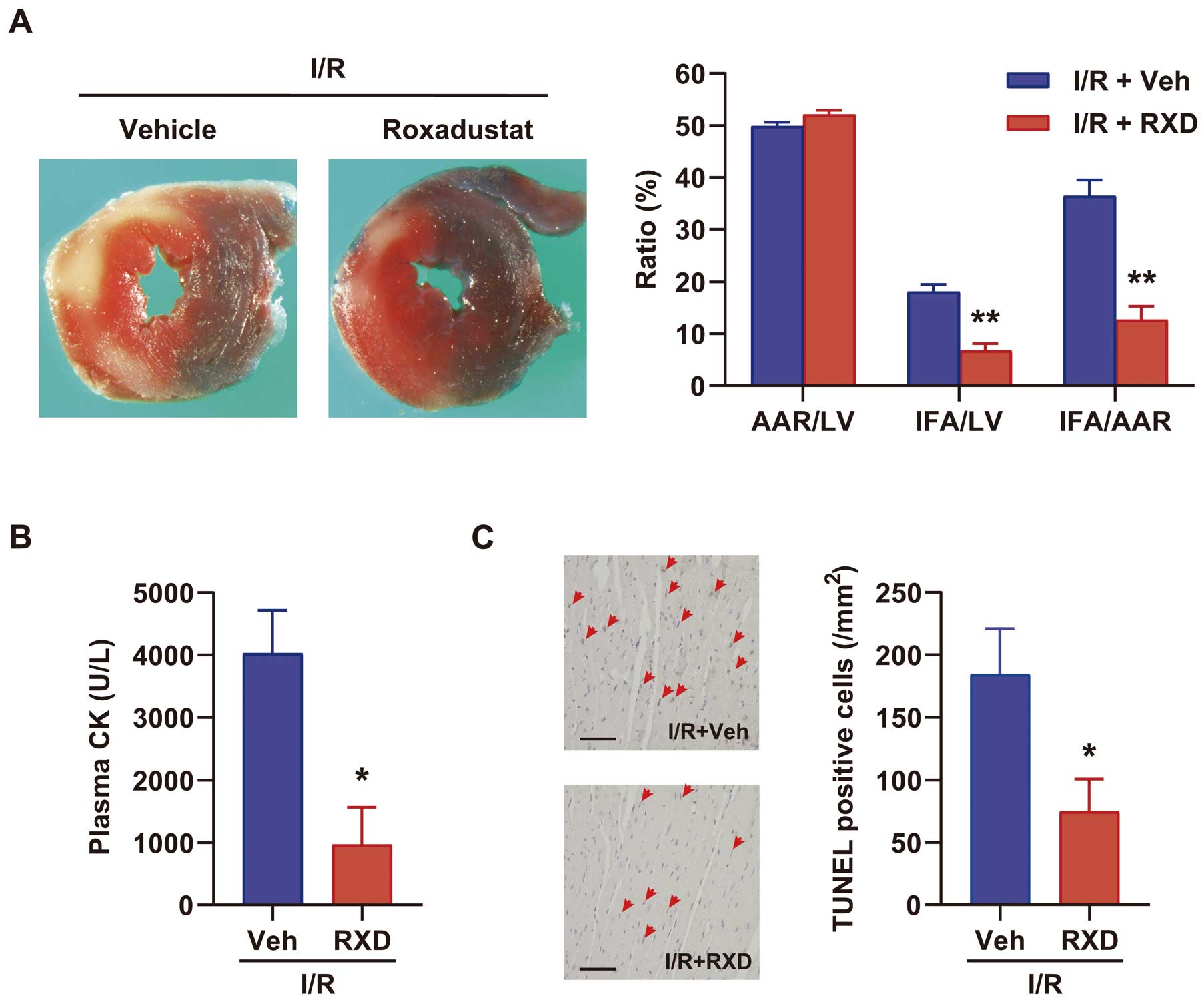
at 3 h (Figure 1B), and steadily increased Hif-1α-regulated proteins in the myocardium such as PKM2, REDD1 and LDH at 4 h (Figure 1C). RXD pretreatment (25 mg/kg, 4 h before LAD ligation) markedly reduced the infarct size, characterized by infarct area (IFA) and IFA per area at risk (IFA/AAR) at 3 h after reperfusion (Figure 2A). It also significantly suppressed plasma CK activity (Figure 2B) and reduced TUNEL-positive cells, a marker of programmed cell death, in the injured myocardium 6 h after reperfusion (Figure 2C).
To identify the potential mechanism underlying RXD-induced ischemic tolerance, we examined OCR and ECAR in cultured cardiomyocytes under RXD treatment. RXD (25 μmol/L, approximately corresponding to Cmax
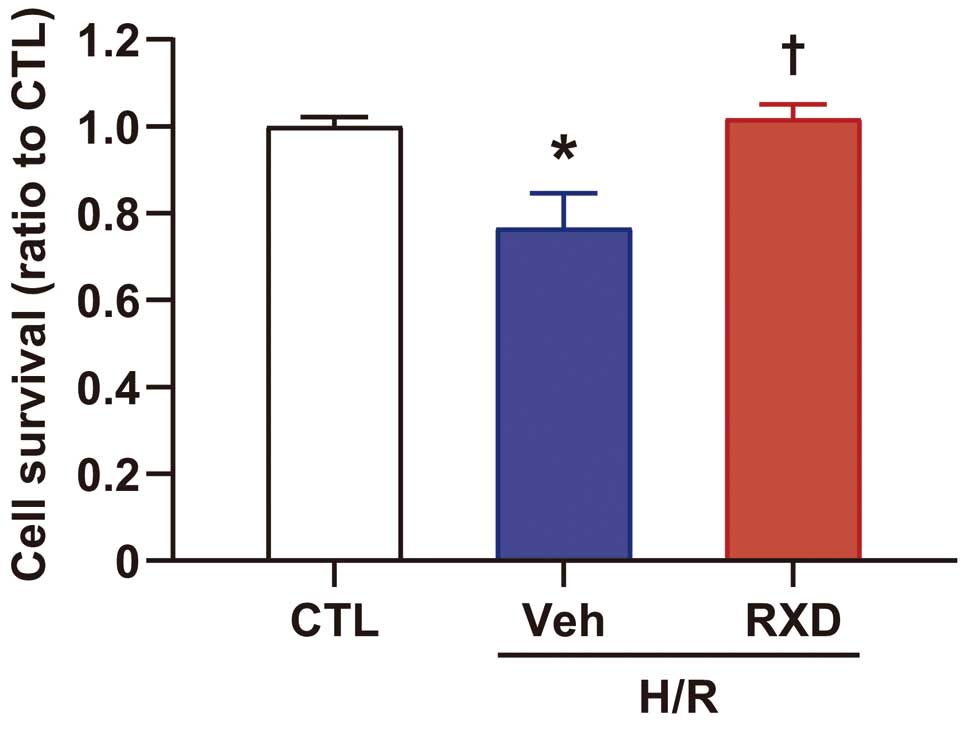
after single oral administration (1‒2 mg/kg) in humans) markedly enhanced Hif-1α expression in cultured cardiomyocytes (Figure 3A). XFp analysis showed that OCR was significantly suppressed (Figure 3B,C), and ECAR was significantly enhanced after RXD treatment (Figure 3D,E). Conversely, silencing Hif-1α abolished the enhancement of ECAR, but not the reduction of OCR. These results suggested that RXD enhanced anaerobic respiration through Hif-1α and reduced OCR independently of Hif-1α. Finally, we found that pretreatment with RXD significantly prevented cell death induced by H/R (Figure 4). Taken together, these results showed that RXD pretreatment was able to produce ischemic tolerance by shifting metabolism from aerobic to anaerobic respiration, and that it was able to maintain the ATP production without oxygen, resulting in survival of cardiomyocytes after H/R.

Discussion
Hif-1α is a pivotal transcriptional factor in the physiology and pathophysiology of the cardiovascular system;15
Semenza et al first identified Hif-1α as a transcriptional factor for erythropoietin,16
and Ratcliffe and Kaelin demonstrated the mechanism of Hif-1α degradation by VHL in normoxia and Hif-1α activation in hypoxia.17,18
In particular, numerous studies suggest that PHD inhibition or Hif-1α overexpression reduces ischemic injury.15
Here, we demonstrated that RXD, which is being adopted into clinical practice, markedly reduced I/R injury by pharmacologically utilizing hypoxic responses through PHD inhibition. Given that several clinical trials have currently failed to show clinical benefits from IPC or RIPC, our findings suggested that pharmacological preconditioning with RXD might be a feasible and novel strategy to further reduce myocardial ischemic injury.
In this study, we also showed that RXD enhanced anaerobic respiration and suppressed aerobic respiration, resulting in a respiratory metabolic shift. Mechanistically, silencing Hif-1α abolished the enhancement of anaerobic respiration, but not the reduction of aerobic respiration. These results suggested that anaerobic respiration is enhanced in a Hif-1α-dependent manner, whereas Hif-1α was not responsible for the acute response of OCR reduction at 4 h after RXD treatment. As it was recently reported that there are various substrates targeted by PHDs, PHDs may hydroxylate other proteins, excluding Hif-1α, and partly mediate hypoxic responses independently of Hif-1α.
Furthermore, we demonstrated that RXD pretreatment significantly prevented cell death induced by a sudden induction of hypoxia following reoxygenation. This might be because production of oxygen-independent ATP through enhanced anaerobic respiration can maintain myocytes under ATP-starved conditions, conferring cardioprotection against ischemic injury. Also, reduced oxygen consumption may contribute to suppressing oxidative stress after reoxygenation, resulting in cardioprotection against reoxygenation injury. Nevertheless, it will be difficult to identify the simple molecular mechanism by which Hif-1α produces ischemic tolerance because Hif-1α regulates the expression of numerous genes. Therefore, we deduced that many molecular mechanisms, including metabolic shift, might cooperatively work to produce ischemic tolerance under RXD treatment.
On the other hand, pretreatment before ischemia would be required because of the time needed to upregulate Hif-1α-regulated genes to achieve effective cardioprotection. Hence, pharmacological preconditioning with RXD before elective CABG will be a suitable clinical application. Furthermore, cardioprotection during heart transplantation might be another application, as has been shown for liver transplantation.20
In conclusion, RXD markedly reduced the infarct size in I/R injury. Pharmacological preconditioning with RXD may be a novel strategy against myocardial I/R injury.
Acknowledgments
We thank Midori Sato and Akiko Hanada for their excellent experimental and technical assistance.
Funding
This work was supported by JSPS KAKENHI grants (M. Ikeda: 16H07049 and 18K15892, T. Ide: 17K09582, H. Tsutsui: 15H04815), and research grants from the Takeda Science Foundation (M. Ikeda); the Uehara Memorial Foundation (M. Ikeda); the Japan Foundation for Applied Enzymology (VBIC: Vascular Biology of Innovation) (M. Ikeda); YOKOYAMA Foundation for Clinical Pharmacology (M. Ikeda, YRY-1911); MSD Life Science Foundation, Public Interest Incorporated Foundation (M. Ikeda); and the AMED (grant no. 19ek0109339 h0002: H. Tsutsui).
Conflicts of Interest
H. Deguchi, T. Tadokoro, S. Ikeda, K. Okabe, A. Ishikita, and S. Matsushima have nothing to declare. K. Saku received a consultant fee from Abiomed Japan K.K. H. Tsutsui received a consultant fee from Nippon Boehringer, Bayer Yakuhin, Novartic Pharma, and Ono Pharmaceutical. H. Tsutsui received remuneration from MSD K.K., Astellas Pharma Inc., Pfizer Japan Inc., Bristol-Myers Squibb Company, Otsuka Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Mitsubishi-Tanabe Pharma Corporation, Nippon Boehringer Ingellheim Co., Ltd., Takeda Pharmaceutical Company Limited, Bayer Yakuhin, Ltd., Novartis Pharma K.K., Kowa Pharmaceutical Co. Ltd., and Teijin Pharma Ltd. H. Tsutsui received a manuscript fee from Medical View and Nippon Rinsho. K. Saku received research funding from Omron Healthcare Co. H. Tsutsui received research funding from Nippon Boehringer, Mitsubishi-Tanabe Pharma, Japan Tobacco, Daiichi Sankyo, IQVIA Services Japan, Acterion Pharmaceuticals Japan, and Omron Healthcare. M. Ikeda received scholarship funds from Takeda Science Foundation, the Uehara Memorial Foundation, the Japan Foundation for Applied Enzymology (VBIC: Vascular Biology of Innovation), the YOKOYAMA Foundation for Clinical Pharmacology (YRY-1911), MSD Life Science Foundation, and the Public Interest Incorporated Foundation. H. Tsutsui has an endowed department by Acterion Pharmaceuticals Japan.
References
- 1.
He J, Ogden LG, Bazzano LA, Vupputuri S, Loria C, Whelton PK. Risk factors for congestive heart failure in US men and women: NHANES I epidemiologic follow-up study. Arch Intern Med 2001; 161: 996–1002.
- 2.
Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, et al. Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: A randomised controlled trial. Lancet 2007; 370: 575–579.
- 3.
Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, et al. Remote ischemic preconditioning and outcomes of cardiac surgery. N Engl J Med 2015; 373: 1408–1417.
- 4.
Meybohm P, Bein B, Brosteanu O, Cremer J, Gruenewald M, Stoppe C, et al. A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med 2015; 373: 1397–1407.
- 5.
Hausenloy DJ, Kharbanda RK, Moller UK, Ramlall M, Aaroe J, Butler R, et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019; 394: 1415–1424.
- 6.
Hausenloy DJ. Cardioprotection techniques: Preconditioning, postconditioning and remote conditioning (basic science). Curr Pharm Des 2013; 19: 4544–4563.
- 7.
Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, et al. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc Res 2008; 77: 463–470.
- 8.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148: 399–408.
- 9.
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol Cell 2008; 30: 393–402.
- 10.
Villa JC, Chiu D, Brandes AH, Escorcia FE, Villa CH, Maguire WF, et al. Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell Rep 2014; 8: 1077–1092.
- 11.
Minamishima YA, Kaelin WG Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science 2010; 329: 407.
- 12.
Provenzano R, Besarab A, Wright S, Dua S, Zeig S, Nguyen P, et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: A phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am J Kidney Dis 2016; 67: 912–924.
- 13.
Fujino T, Ide T, Yoshida M, Onitsuka K, Tanaka A, Hata Y, et al. Recombinant mitochondrial transcription factor A protein inhibits nuclear factor of activated T cells signaling and attenuates pathological hypertrophy of cardiac myocytes. Mitochondrion 2012; 12: 449–458.
- 14.
Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc 2014; 9: 421–438.
- 15.
Bishop T, Ratcliffe PJ. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ Res 2015; 117: 65–79.
- 16.
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci USA 1991; 88: 5680–5684.
- 17.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399: 271–275.
- 18.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2000; 2: 423–427.
- 19.
Xie L, Pi X, Townley-Tilson WH, Li N, Wehrens XH, Entman ML, et al. PHD2/3-dependent hydroxylation tunes cardiac response to beta-adrenergic stress via phospholamban. J Clin Invest 2015; 125: 2759–2771.
- 20.
Zhang X, Liu Z, Xiao Q, Zeng C, Lai CH, Fan X, et al. Donor treatment with a hypoxia-inducible factor-1 agonist prevents donation after cardiac death liver graft injury in a rat isolated perfusion model. Artif Organs 2018; 42: 280–289.