Reviews
Lipoprotein(a) and Cardiovascular Diseases ― Revisited ―
2020 年 84 巻 6 号 p. 867-874

詳細
2020 年 84 巻 6 号 p. 867-874
Two decades ago, it was recognized that lipoprotein(a) (Lp(a)) concentrations were elevated in patients with cardiovascular disease (CVD). However, the importance of Lp(a) was not strongly established due to a lack of both Lp(a)-lowering therapy and evidence that reducing Lp(a) levels improves CVD risk. Recent advances in clinical and genetic research have revealed the crucial role of Lp(a) in the pathogenesis of CVD. Mendelian randomization studies have shown that Lp(a) concentrations are causal for different CVDs, including coronary artery disease, calcified aortic valve disease, stroke, and heart failure, despite optimal low-density lipoprotein cholesterol (LDL-C) management. Lp(a) consists of apolipoprotein (apo) B100 covalently bound to apoA. Thus, Lp(a) has atherothrombotic traits of both apoB (from LDL) and apoA (thrombo-inflammatory aspects). Although conventional pharmacological therapies, such as statin, niacin, and cholesteryl ester transfer protein, have failed to significantly reduce Lp(a) levels, emerging new therapeutic strategies using proprotein convertase subtilisin-kexin type 9 inhibitors or antisesnse oligonucleotide technology have shown promising results in effectively lowering Lp(a). In this review we discuss the revisited important role of L(a) and strategies to overcome residual risk in the statin era.
Cardiovascular disease (CVD) is a leading cause of mortality worldwide. Although statins markedly improve cardiovascular (CV) outcomes by lowering low-density lipoprotein cholesterol (LDL-C), CV events persist in a substantial proportion of patients.1 Worldwide, efforts to abrogate CV events in such patients (i.e., those with residual CV risk) have been made by modulating non-LDL-C, such as triglyceride-rich lipoprotein cholesterol or lipoprotein(a) (Lp(a)).2–5
Lp(a), an LDL-like particle synthesized in the liver, is known to be elevated in patients with CV events despite optimal LDL-C management.6 Lp(a) consists of apolipoprotein (apo) B100 covalently bound to apoA. Lp(a) characteristically inherits atherogenicity from both apoB and apoA, as well as prothrombogenic and proinflammatory traits from apoA. This collectively confers CVD-prone traits, unlike other lipoprotein particles that only have apoB (Figure).
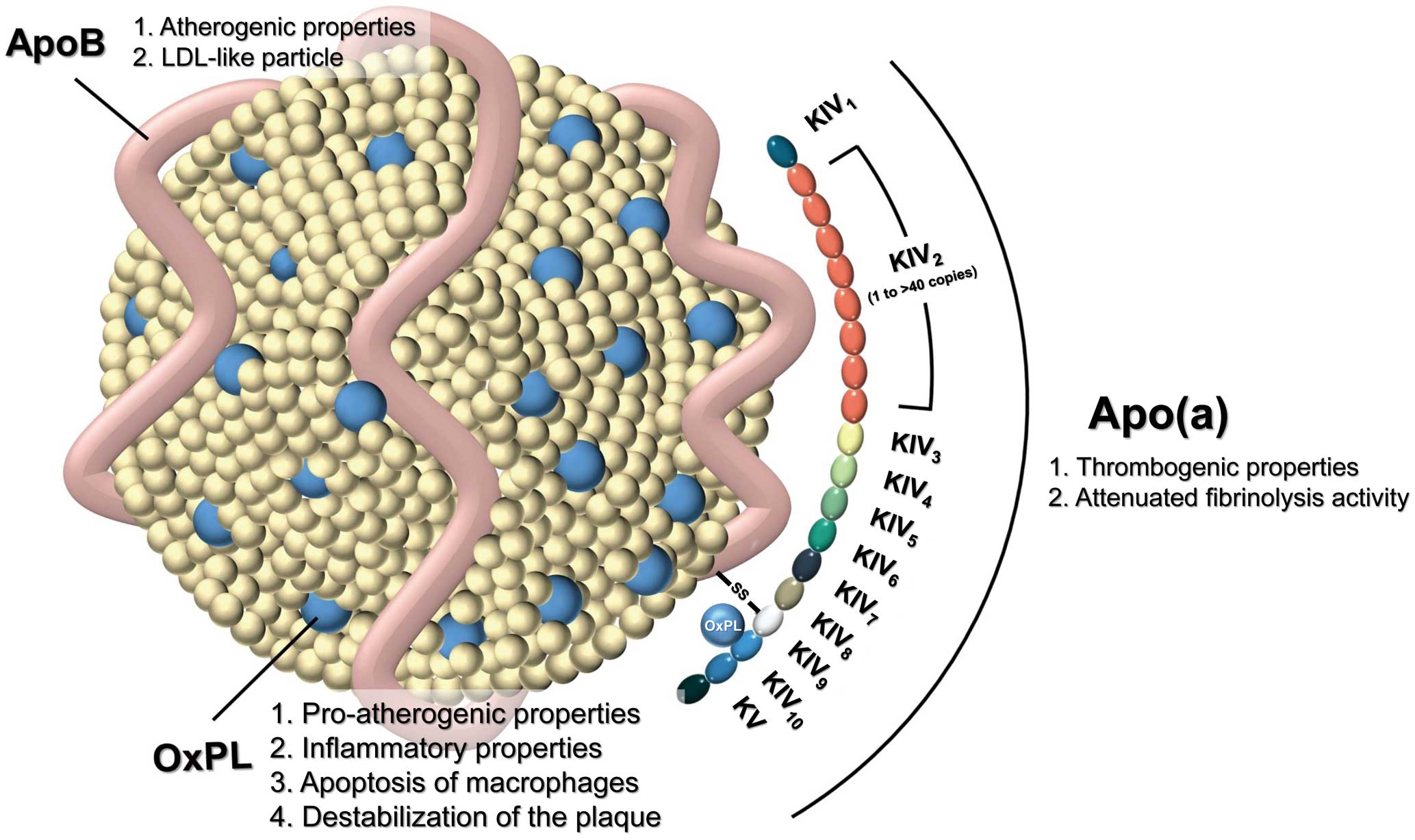
Schematic illustration of lipoprotein(a) (Lp(a)). Lp(a) consists of apolipoprotein (apo) B covalently bound to (apoA), which itself consists of a structure called kringle domains (K), which share structural homology with plasminogen. ApoA contains KIV and KV, with KIV having evolved 10 types of duplications, of which only KIV2 has expanded copy numbers (ranging from 1 to > 40 copies, depending on the individual) Additional oxidized phospholipid (OxPL) is bound covalently to KIV10, as well as in lipid phase of the low-density lipoprotein (LDL) particle.
These atherogenic, prothrombogenic, and proinflammatory traits led to the “Lp(a) hypothesis”, namely that CV outcomes would be reduced by lowering Lp(a) concentrations. Although many attempts have been made to test the Lp(a) hypothesis over the past 2 decades, until recently no intriguing Lp(a)-lowering therapeutics had reached the randomized trial stage. However, novel technologies, such as antisense oligonucleotides (ASO), have enabled investigation as to whether Lp(a) is a critical contributor to residual CV risk.7,8 In this article, we review the properties of Lp(a) that confer susceptibility to CVDs and the clinical trials that led to the development of the most up-to-date therapeutics.
Apo(a) has structural homology to plasminogen because of a shared structure called the kringle domain, a triple loop stabilized by 3 disulfide bonds.9 Plasminogen is characterized by 5 different kringle domains (Kringle [K] I-KV), of which only KIV and KV are present in the Lp(a)-encoding LPA gene. The LPA gene has 10 different types of duplicated KIV domains, whereas the KI–KIII domains are deleted. Of the 10 types of KIV domains, KIV type 2 (KIV2) has expanded copy numbers (from 1 to >40 copies), whereas KIV1 and KIV3 –KIV10 are present as single copies (Figure). Additional oxidized phospholipids (OxPL) are bound covalently to KIV10, as well as in the lipid phase of the LDL particle. The number of KIV2 copies varies among individuals, resulting in an isoform size polymorphism of apoA. The molecular weight of apoA is determined by the number of kringle domains. The phenotype of the number of kringle domains in individuals depends on the genetic information inherited from each parent. Other important determinants of the number of kringle domains are single nucleotide polymorphisms (SNPs), which are a single mutations of nucleotides at a specific position of a certain gene that result in different traits of the gene.9
Lp(a) levels in human samples are mainly measured using immunoassays with polyclonal antibodies against apoA.10 The antibodies used in such assays recognize the kringle domains, although they cannot specify which domain in which kringle domain. Because the assay measures the total amount of kringle domains, Lp(a) levels may be under- or overestimated in patients with small (fewer kringle domains) or large (more kringle domains) apoA isoforms, respectively. These issues can be improved by using standardized calibration and expressing Lp(a) levels as the concentration of apoA particles (nmol/L).10
The risk of coronary artery disease (CAD) increases with increasing Lp(a) concentrations.6 In a pooled cohort of 126,634 patients, CAD risk was found to increase by 16% with every 1SD increase in Lp(a).11 The Copenhagen City Heart Study revealed that the 95th percentile group had a 2.6-fold higher risk of CAD than the 22th percentile group.12 This phenomenon is still observed after adjusting for the number of KIV2 domains, suggesting that Lp(a) concentration and the number of KIV2 domains are independently associated with the risk of CAD.13 A prospective study of 15-year outcomes of patients also revealed that CVD, defined as a composite of vascular death, acute CAD, and ischemic stroke, was 2.34-fold higher for patients in the top quintile than for those in the other quintiles.14 In a large contemporary general population study, high plasma Lp(a) concentrations were associated with increased risk of ischemic stroke, both observationally and causally from human genetics.15 The risk of heart failure also increases by 1.54-fold for high compared with low Lp(a).16 Higher Lp(a) concentrations were associated with an increased risk of progressive coronary atherosclerosis and acute myocardial infarction (MI), which had an especially high population burden in South Asians and Latin Americans.17–19 Due to such high CVD risk associated with elevated Lp(a) levels, recent lipid guidelines stress the importance of evaluating Lp(a) levels to stratify CVD risk.20,21
Unlike other lipoproteins, genetics have a greater effect on Lp(a) concentrations than individual lifestyle factors. A genetic study using the Mendelian randomization approach in multi-ethnic groups demonstrated a strong relationship between Lp(a) concentration and CVD risk.22 Patients across all ethnic groups with a polymorphism that had smaller apoA but a high Lp(a) concentration were associated with a higher risk of CVD.22 One of the causes of such genetic traits are SNPs of the LPA gene. Although there is no evidence that SNPs themselves modulate Lp(a) concentrations, they are associated with small isoforms, which do mediate high levels. Certain SNPs, such as rs3798220 and rs10455872, are associated with a lower number of KIV2 domains and hence small apoA isoforms, which are inversely related to Lp(a) concentrations.23 These SNPs are also associated with elevated odds ratios for CAD. Remarkably, when both SNPs are present in a single person, the odds ratio for CAD increases over 4-fold, which is higher than any other conventionally known CAD risk factor.23
The association between Lp(a) and CAVD has recently been reported.24 Oxidized lipids are major factors that cause vascular inflammation in atherosclerosis. Previous studies demonstrated a positive association between elevated Lp(a) concentrations and increased risk of CAVD in prospective epidemiologic studies.25 Genetic variation in the Lp(a) locus, which affects Lp(a) concentrations, is correlated with aortic valve calcification in many ethnic populations, as well as with incident clinical aortic stenosis.
Pathologic studies of vulnerable human coronary and carotid plaques showed differential expression of oxidation-specific epitopes and apoA when plaques progress, rupture, and become clinically symptomatic.26 Explanted human valves showed increased Lp(a) and OxPL content, which may mediate adverse CV outcomes during treatment.27 Phosphocholine-containing OxPL may play an important role in CAVD. Recently, elevated Lp(a) and OxPL-apoB levels has been associated with faster aortic stenosis aggravation and the need for aortic valve replacement.28 These findings support the hypothesis that Lp(a) mediates aortic stenosis progression via its associated OxPL. Thus, future randomized trials of Lp(a)- and OxPL-apoB-lowering therapies in aortic stenosis are needed.24
Lp(a) and OxPL may induce CAVD by binding to exposed or denuded valve surfaces through potent lysine-binding sites. The OxPL of Lp(a) is subsequently converted into procalcifying lysophosphatidic acid via the enzyme autotaxin, which promotes inflammation and fibrosis.29 A case-control study showed that patients with CAVD had higher levels of Lp(a) OxPL-apoB, and autotaxin, which suggests that the Lp(a) provides OxPL and autotaxin in the valvular cells to further induce inflammation and calcification.30 In summary, the effects of OxPL and autotaxin activity, incorporated in Lp(a), may contribute to CAVD development, which is characterized by stages of lipid deposition, inflammation, fibrosis, calcification, and eventually symptomatic stenosis.24
The structural combination of apoB-containing LDL-C and apoA confers the superior atherothrombogenicity of Lp(a) over apoB-only LDL-C. The lysine-binding site of the kringle domains within an apoA molecule predisposes Lp(a) molecules to bind to the endothelial receptors, thereby contributing to atherogenicity. The structural homology of apoA to the plasminogen molecule also confers thrombogenicity of Lp(a). Therefore, an excess concentration of Lp(a) abrogates the function of plasmin activators, decreases plasmin levels, and eventually leads to attenuated fibrinolysis activity.31
OxPL plays a major role in the proatherogenic and inflammatory properties of the Lp(a) molecule. OxPL is mainly an oxidized form of polyunsaturated fatty acyl chains of phospholipid.32 OxPL is recognized by pattern recognition receptors on innate immune cells. The primed innate immune cells then recruit other circulating immune cells by secreting cytokines such as interleukin (IL)-1, IL-6, and IL-8, which further evokes nuclear factor (NF)-κB signaling to accelerate the immune process.27 Pattern recognition receptors such as Toll-like receptor 2 and CD36 were shown to recognize OxPL to trigger apoptosis of macrophages, which leads to necrosis of the atherosclerotic plaque and ultimately results in destabilization of the plaque.33 These findings may collectively explain the increased levels of OxPL in patients with acute coronary syndromes or carotid/femoral artery atherosclerosis and the association with MI and stroke.34
The proatherogenic, inflammatory, and antifibrinolytic properties of the Lp(a) molecule are summarized in Figure.
The metabolic pathways of Lp(a) are not fully understood. Levels of Lp(a) are known to be determined by the production of Lp(a) via LPA gene expression and clearance, which are influenced by the APOE gene, low-density lipoprotein receptor (LDLR) and LDLR-related protein 1 (LRP1).35–37 A recent meta-analysis indicated an 11% increase in Lp(a) in statin-treated groups. An in vitro study demonstrated that the increase in Lp(a) levels in statin-treated hepatocytes was accounted for by elevated LPA gene expression stimulated by the use of statin.36 Another report suggested that the Lp(a)-reducing effect of proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibodies (mAbs) may be mediated by very low-density lipoprotein and apoE.37
Therapeutic lifestyle changes (TLCs) have been shown to be effective treatment strategies for both primary and secondary prevention of CVD risk.
Dietary interventions have failed to demonstrate a marked change in Lp(a) levels, as indicated in Table 1. Because it has been shown that a healthy lifestyle lowers the risk of CVD, many dietary randomized trials were conducted to demonstrate that healthy dietary interventions, such as a low-fat diet, may reduce Lp(a) levels. Interestingly, however, these expectations were contradicted by the results of the trials. A low-fat, high-vegetable diet, which is conventionally considered as a diet to lower CVD risk, resulted in higher Lp(a) levels that a low-fat, low-vegetable diet.38,39 In addition, in the Dietary Approaches to Stop Hypertension (DASH) study, the experimental arm with more unsaturated fat showed higher Lp(a) levels than the high-protein or -carbohydrate arms.40 These findings are intriguing, because unsaturated fats are conventionally demonstrated to lower other lipid profiles, such as LDL-C. A more comprehensive CV health-diet metric, defined as body mass index, healthy diet score, daily fish consumption, whole grain consumption, low sodium intake, and low sweetened beverage consumption, demonstrated a modest effect on Lp(a). However, a healthier dietary metric showed substantial improvement in CVD outcomes.41
| Reference | Year | Type of trial | Sample | Type of intervention | Result |
|---|---|---|---|---|---|
| 42 | 1994 | Multicenter prospective study |
n=2,500 (Finnish young adults, 9–24 years of age) |
Physical activity assessed by questionnaires |
Significant inverse correlation between Lp(a) and physical activity |
| 43 | 1994 | Cross-sectional study | n=150 (100 men, 50 women; mean age 44 and 35 years, respectively) |
Physical capacity | No significant association between Lp(a) and exercise capacity |
| 44 | 2001 | Cross-sectional, cross- cultural study |
n=140 (40–70 years of age; African American, Native American, and Caucasian) |
Moderate, moderate-vigorous physical activity levels, and total MET-min/day |
No significant association between Lp(a) and physical activity or maximal treadmill time |
| 38 | 2004 | Randomized crossover design |
n=36 (healthy women) | Low-fat, low-vegetable and low-fat, high-vegetable diet crossover |
Low-fat, high-vegetable diet showed higher Lp(a) levels |
| 39 | 2010 | Randomized crossover design |
n=63 (men and women, >20 years of age) |
LFHC or HFLC diet | LFHC diet-induced increase in Lp(a) associated with increase OxPL and small LDL |
| 40 | 2014 | Randomized crossover design (The Omni Heart Trial) |
n=155 (89 Blacks, 66 Whites) |
DASH-type healthy diets rich in carbohydrates, protein, or unsaturated fat |
DASH diets high in unsaturated fat increased Lp(a) levels less than protein- or carbohydrate- rich DASH diets |
| 41 | 2016 | Population-based cohort (The EPIC-Norfolk prospective population study) |
n=25,639 (men and women between 39 and 79 years of age) |
Stratification by CV health metrics (BMI, healthy diet score, daily fish, and whole grain, low sodium, and low sweetened beverage consumption) |
Lp(a) levels were not markedly affected by CV health metrics, although CV disease risk was substantially reduced |
BMI, body mass index; CV, cardiovascular; DASH, Dietary Approaches to Stop Hypertension; HDL, high-density lipoprotein; HFLC, high-fat, low-carbohydrate diet; LDL, low-density lipoprotein; LFHC, low-fat, high-carbohydrate diet; Lp(a), lipoprotein(a); MET, metabolic equivalent; OxPL, oxidized phospholipid.
The association between physical activity and Lp(a) levels has also been studied (Table 1). An inverse correlation was shown between physical activity and Lp(a).42 In another study, performed on 150 Caucasians (100 men, 50 women), Lp(a) concentrations were not significantly associated with exercise capacity, age, sex, body composition, or other lipoproteins.43 Other cross-sectional studies also failed to demonstrate an association between physical activity and Lp(a).44 Although inconclusive, these results collectively fail to show an association between Lp(a) and regular physical activity.
Collectively, this evidence shows that TLC, including dietary and exercise interventions, is not associated with favorable serum Lp(a) concentrations. The lack of effect of dietary or exercise interventions on serum Lp(a) levels points to the importance of the genetic factors that predispose to high Lp(a) levels. Thus, novel therapeutic strategies to lower Lp(a) levels are crucial.
Pre-existing lipid-modulating agents have been tested for their Lp(a)-lowering effects and CV outcomes. A meta-analysis regarding the effect of statins on Lp(a) levels and subsequent CV outcomes has been published.45 Statins showed benefits on conventional CV risk factors, although a modest increase was noted in Lp(a) levels. Interestingly, however, poor CV outcomes were significantly more strongly associated with on-statin than placebo-allocated patients, where high (≥50 mg/dL) Lp(a) concentrations were noted at baseline and during the follow-up period. These populations with high Lp(a) concentrations may account for the residual CV risk despite statin therapy. In this population with elevated Lp(a) concentrations, therapeutic agents targeting Lp(a) may mitigate Lp(a)-mediated CV risk.2
Therapeutics with Lp(a)-lowering effects have been tested for their CV benefits (Table 2). A seminal study showed that performing apheresis to lower LDL-C in patients with CV disease and Lp(a) greater than the 95th percentile had promising effects in decreasing CV events. The marked reduction in Lp(a) concentrations was associated with a substantial drop in annual MI (97%) and composite CV outcomes (86%) rates.46 These promising results were followed by a multicenter observational study that prospectively evaluated 170 high-risk patients with mean LDL-C and Lp(a) concentrations of 99.0 and 104.9 mg/dL, respectively.47 Intriguingly, rates of annual composite CV outcomes (−78%), MI (−85.7%), and revascularization through either percutaneous coronary intervention (−68.2%) or coronary artery bypass grafting (−80%) were reduced.47 These data are the first to show that, in high-risk patients who undergo apheresis, the reduction in Lp(a) is associated with improved CV outcomes. These results further supported the rationale behind lowering Lp(a) concentrations to decrease CVD risk, although such results have not been replicated and have only been presented in small sample studies.
| Reference | Year | Type of intervention |
Type of trial | Sample | Baseline Lp(a) concentration |
Lp(a) reduction (%) |
CV benefit |
|---|---|---|---|---|---|---|---|
| 46 | 2009 | Lipid apheresis | Longitudinal, multicenter, cohort study |
n=120 (patients with maximal lipid-lowering therapy and high CVD risk, Lp(a) >2.14 μmol) |
4.21 μmol/L | 73.0 | Yearly MACE rates were decreased by 86% |
| 47 | 2013 | Lipid apheresis | Longitudinal, multicenter, cohort study |
n=170 (patients with maximal lipid-lowering therapy and high CVD risk, Lp(a) >2.14 μmol) |
3.95 μmol/L | 66.6 | Yearly MACE, MI, PCI, CABG rates were decreased by 60–86% |
| 48 | 2013 | Niacin plus simvastatin vs. simvastatin plus placebo (target: LDL 40–80 mg/dL) |
AIM-HIGH trial (prospective, randomized, double-blind, placebo-controlled trial) |
n=3,414 | 33.8 nmol/L (median) |
19.0 | Not associated with CV benefit |
| 52 | 2010 | Anacetrapib (CETP inhibitor) vs. placebo |
Randomized, double-blind study, Phase 3 trial |
n=15,067 (patients with CV risk factors between 18 and 80 years of age who take statins) |
26.8 nmol/L (median) |
36.8 | CV benefit was not shown |
| 53 | 2015 | Randomly allocated to 1 of 9 treatments including TA-8995 (CETP inhibitor), rosuvastatin, or placebo |
Randomized, double-blind, placebo-controlled, parallel-group Phase 2 trial |
n=364 (mild dyslipidemia) |
35–39 nmol/L (median of each dose) |
26.7–36.9 (depending on dose) |
CV outcome benefits are yet to be demonstrated |
| 54 | 2019 | Evolucumab (PCSK9 inhibitor) or placebo s.c. injection every 2 or 4 weeks |
FOURIER trial, randomized, double-blind, placebo-controlled trial (post hoc analysis) |
n=27,654 (patients with established CVD, 40–85 years of age) |
37 nmol/L (median) |
26.9 | Patients with higher baseline Lp(a) had greater absolute reductions of Lp(a) and coronary benefit |
| 55 | 2018 | Randomly allocated to inclisiran (PCSK9 siRNA conjugated to GalNAc) or placebo |
ORION 1 trial, randomized, double-blind, placebo-controlled trial, Phase 2 trial |
n=501 (patients with established CVD or risk equivalents, receiving maximum tolerated statin dose) |
32.0– 47.0 nmol/L (median of each dose) |
14–26 (NS) | There was interindividual response variability in Lp(a) reduction |
| 56 | 2015 | 2:1 allocation to weekly s.c. mipomersen (ASO to apoB-100 or placebo injection for 26 weeks |
Randomized, double-blind, placebo-controlled Phase 3 trial |
n=382 (patients with familial hypercholesterolemia on maximal medical therapy) |
32.0 mg/dL (∼80 nmol/L; median of pooled analysis) |
26.4 (pooled analysis) |
CV outcome benefits are yet to be demonstrated |
| 8 | 2016 | Randomly allocated to IONIS-APO(a)Rx (ASO targeting apoA) or placebo |
Randomized, double-blind, placebo-controlled, dose-titration Phase 2 trial |
n=64 (Cohort A: Lp(a) 125–437 nmol/L; Cohort B: Lp(a) >438 nmol/L) |
Cohort A (>80th percentile): 261.4 nmol/L Cohort B (>99th percentile): 457.6 nmol/L |
Cohort A: 62.8 vs. placebo Cohort B: 67.7 vs. placebo |
N/A |
| 8 | 2016 | Randomly allocated to IONIS- APO(a)Rx-Lrx (ASO targeting apoA bound to GalNac) or placebo |
Randomized, double-blind, placebo-controlled, ascending-dose Phase 1/2a trial |
n=58 (healthy volunteers with Lp(a) >75 nmol/L) |
111.4– 196.8 nmol/L (median of each ascending dose group) |
Mean reduction: 92.4 |
N/A |
| 63 | 2020 | Randomly allocated to different doses and dosing intervals of IONIS- APO(a)Rx-Lrx |
Randomized, double-blind, placebo-controlled, dose-ranging trial |
n=286 (patients with established CVD with Lp(a) >60 mg/dL [150 nmol/L]) |
224.3 nmol/L (median of pool population) |
20-mg monthly dose: 35 20-mg weekly dose: 80 Control: 6 |
N/A |
apo, apolipoprotein; ASO, antisense oligonucleotide; CABG, coronary artery bypass graft; LDL, low-density lipoprotein; CETP, cholesterylester transfer protein; CV, cardiovascular; CVD, cardiovascular disease; GalNAc, N-acetylgalactosamine; MACE, major adverse cardiac events; MI, myocardial infarction; N/A, not available; PCI, percutaneous coronary intervention; PCSK9, proprotein convertase subtilisin/kexin type 9; siRNA, short interference RNA.
Extended-release niacin in addition to statin therapy was studied in the AIM-HIGH trial.48 However, in that trial the modest decrease in Lp(a) of 19% in the niacin plus statin combined therapy group compared with the placebo group did not result in a reduction in CV events, possibly because the study was insufficiently powered. Whether niacin will reduce CV risk remains unknown because randomized control trials with adequate power for Lp(a) and CV event rates have not been conducted. Originally developed as an agent to increase high-density lipoprotein (HDL), cholesteryl ester transfer protein (CETP) inhibitors have also exhibited LDL-C and Lp(a) lowering effects. Randomized trials of 3 of the 5 CETP inhibitors, namely torcetrapib, evacetrapib, and dalcetrapib, were prematurely terminated either for toxicity or futility.49–51 Data regarding Lp(a) were published in trails of anacetrapib and TA-8995, although the decrease in Lp(a) of 20–40% was not associated with CV benefit.52,53
The Lp(a)-lowering effect of PCSK9 mAbs is well established, as demonstrated in the FOURIER trial. Evolocumab reduced Lp(a) by 26.9% independent of baseline LDL-C concentrations. Interestingly, patients with Lp(a) concentrations above the median (>37 nmol/L) had a greater rate of absolute risk reduction and number needed to treat compared with the placebo control group by 1.41% and 71, respectively.54 Another PCSK9-targeting agent, inclisiran, which is a small interference RNA that blocks the synthesis of PCSK9, has shown prolonged effects in decreasing LDL-C after a single dose.55 However, in contrast with evolucumab, the effects of inclisiran in reducing Lp(a) failed to reach statistical significance (−14% or −26% with 1 or 2 doses).55 In another study, mipomersen, an ASO targeting apoB, was randomized to patients with familial hypercholesterolemia on maximal medical therapy.56 In patients randomized to 200 mg mipomersen daily, the Lp(a) concentration was reduced by 26.4%, although CV benefits are yet to be demonstrated.56
Although the therapeutic agents described above have proven Lp(a)-reducing effects, their CV benefit is either unproven or absent. A 19% modest reduction in Lp(a) following the use of statin plus extended-release niacin therapy did not exhibit CV benefits.48 CETP inhibitors also reduced Lp(a) by 20–40%, although such drugs are not approved for market because of non-CV adverse effects.52,53 PCSK9 mAbs such as evolucumab reduced Lp(a) by 26.9% with modest coronary benefit.54 Overall, these agents showed modest Lp(a)-lowering effect in the range of 20–40%.
In order to achieve a clinically relevant CV benefit, a much larger decrease in Lp(a) concentrations may be necessary. The results of a Mendelian randomization analysis suggest that the clinical benefit may be proportional to the absolute reduction in Lp(a) concentration, claiming that reducing Lp(a) levels by 80–90% in individuals with an Lp(a) concentration above 90–100 mg/dL (225–250 nmol/L) will translate to a 15–20% benefit in CAD events.57 It has been shown that a reduction in LDL-C of 38.67 mg/dL was equivalent to a 101.5-mg/dL (253.8-nmol/L) reduction in Lp(a).57 A more recent Mendelian randomization study proposed that the degree of the reduction in Lp(a) required (101.5 mg/dL) to be equivalent to a decrease in LDL-C of 38.67 mg/dL may have been overestimated due to the influence of SNPs and standardization of the Lp(a) assays used.58 The authors of that study proposed that a 65.7-mg/dL reduction in Lp(a) was comparable to a 38.67-mg/dL reduction in LDL-C.58 Another study evaluated the degree of the reduction in Lp(a) required to improve CV risk in a secondary prevention setting using Mendelian randomization.59 In that study, reductions in Lp(a) of 50 and 99 mg/dL were associated with 20% and 40% decreases in CVD, respectively.59 These results indicate the need for novel therapeutics that reduce Lp(a) by 60–100 mg/dL to provide a CV benefit in contemporary randomized controlled trials.
Currently, there are no drugs approved to lower Lp(a) or randomized trials targeting Lp(a). Lp(a) is not an enzyme or a receptor, such that small molecules are not able to inactivate its function. Because Lp(a) exists at high concentrations within the body, massive amounts of Lp(a)-neutralizing antibodies would be needed to lower Lp(a) concentrations. Such high doses would be expensive and cause adverse immunological reactions.60 Recently, however, promising results were published for 2 clinical trials that used ASO technology to directly inhibit the synthesis of apoA.7,8 One of these trials examined the safety and efficacy of the ASO IONIS-APO(a)Rx (previously ISIS-APO(a)Rx) in subjects with high Lp(a) concentrations.8 Patients were allocated to 2 groups according to Lp(a) concentration: Group A patients were in the >80th percentile (125–437 nmol/L) and Group B patients were in the >99th (>438 nmol/L) percentile. Patients were injected subcutaneously with 100, 200, and 300 mg IONIS-APO(a)Rx once a week for 4 weeks at each dose sequentially. There was a 62.8% and 67.7% decrease in Lp(a) concentrations Group A and Group B, respectively, compared with placebo.8
Despite the enhanced performance of IONIS-APO(a)Rx in lowering Lp(a), a cumulative high dose was required to penetrate into the hepatocytes that synthesize apoA. Such doses are concerning because patients are subjected to potential drug-related toxicity, such as flu-like symptoms and injection site reactions.61 In addition, frequent weekly subcutaneous injections may affect patient compliance. However, by covalently attaching triantennary N-acetylgalactosamine (GalNAc) to the ASO, the drug payload (in this case ASO) can be safely delivered into hepatocytes through an asialoglycoprotein receptor-mediated interaction.62 The second of the ASO technology trials investigated the ASO IONIS-APO(a)Rx-LRx,7 which has a GalNAc covalently linked to the ASO used in the first trial (IONIS-APO(a)Rx) with some minor modifications.62 The GalNAc-conjugation enhanced potency by 30-fold with a mean reduction in Lp(a) concentrations of 92.49%. Tolerability was also improved, because no adverse reactions were observed. In addition, the prolonged effects of IONIS-APO(a)Rx-Lrx made it feasible to inject 80-mg doses once a month or quarter.7 A subsequent randomized double-blind placebo-controlled dose-ranging trial was published recently investigating the reduction in Lp(a) concentrations following the injection of different doses of IONIS-APO(a)Rx-Lrx at different intervals.63 In that study, Lp(a) concentrations were reduced in a dose-dependent manner, with all doses tested achieving a significant reduction. The highest cumulative dose (20 mg weekly) reduced Lp(a) by a mean of 80%.63
These trials are the first to target lowering Lp(a). The ASO technology used in these trials is also in its infancy, although the GalNAc conjugated to the ASO has enabled the drug to effectively lower Lp(a) by up to 99% at a tolerable dose. Previously developed drugs were only able to reduce Lp(a) up to 40%, which did not allow decreases in Lp(a) concentrations to an acceptable range in patients with high Lp(a) concentrations to result in any benefit from the treatment. The results of the Phase 3 outcomes trial of the GalNAc-conjugated ASO therapy that targets Lp(a) synthesis by neutralizing apoA production is likely to be able to test the Lp(a) hypothesis.
It has been shown that high Lp(a) is associated with CVD. The level of Lp(a) is determined genetically and does not respond to therapeutic lifestyle intervention. Although conventional pharmacological therapies, including statins, are not able to mitigate the CV risk elevated by Lp(a), emerging therapeutic strategies using ASO technology have shown promising results in effectively lowering Lp(a). Whether these agents will effectively lower CV risk needs to be investigated in the future.
The authors thank Sotirios Tsimikas (Division of Cardiovascular Diseases, University of California San Diego, La Jolla, CA, USA) for constructiuve comments.
This work was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry for Health and Welfare, Korea (HI15C0987 and HI14C1135) and the Korean Society of CardioMetabolic Syndrome.
K.K.K. holds a patent certificate (10-1579656; pravastatin+valsartan). K.K.K. is an International Associate Editor of the Circulation Journal Editorial Team.