Aging Modulates the Substrate and Triggers Remodeling in Atrial Fibrillation
論文ID: CJ-17-0242
この記事には本公開記事があります。

詳細
論文ID: CJ-17-0242
Aging plays a critical role in the genesis of atrial fibrillation (AF) and also increases the risks of cardiac dysfunction and stroke in AF patients. AF is caused by increased AF triggering from abnormalities of the thoracic vein and/or modulated substrate (atrial) with enhancement of AF maintenance. Clinical and laboratory evidence indicates that aging is significant in the creation of atrial electrical and structural remodeling that leads to increased susceptibility to AF occurrence. Aging is commonly associated with cardiovascular comorbidities, oxidative stress, calcium dysregulation, atrial myopathy with apoptosis, and fibrosis, which all contribute to the genesis of AF. This review updates the current understanding of the effects of aging on the pathophysiology of AF.
The prevalence and severity of arrhythmias increase with age. Advancing age is independently associated with the prevalence of atrial fibrillation (AF).1 Aging and aging-related underlying diseases frequently result in structural remodeling as cardiac morphological changes either grossly or at the histological and ultrastructural levels, or they induce electrophysiological remodeling with changes in cardiac electrical properties in response to a functional insult, or as a result of a structural alteration. Aging-associated cardiac anatomical and functional remodeling may enhance the occurrence or the persistence of AF.
Ectopic impulses generated from the pulmonary veins (PVs) play a key role in the genesis and maintenances of AF.2 Elimination of PV arrhythmogenic foci or targeting atrial substrate abnormalities reduces the AF burden or cures AF.2 PVs contain complex electrical activity because of their distinctive anatomical and physiological characteristics.3 Aging-associated electrophysiological or structural changes in the atria or PVs may facilitate AF occurrence.3 Age is an independent predictor of AF recurrence in lone AF patients after undergoing the first circumferential PV isolation.4 AF may coexist with cardiovascular disorders, which may potentiate its occurrence, and these are also more prevalent with increased age. Together with aging-related myocardial degeneration or anatomical changes, both inflammation and oxidative stress, as consequences of aging, enhance the occurrence and maintenance of AF. The aim of this review is to update the understanding of how aging and AF are connected and highlight the potential role of aging in AF substrate and triggered remodeling.
Structural remodeling is characterized by histopathological changes, such as cellular hypertrophy, apoptosis, or myocardial fibrosis, and anatomical changes, such as chamber dilatation.5
Anatomical Changes With Dilation of PV and Atria in AgingThe left atrium (LA) anteroposterior diameter and wall thickness increase with aging, and the 4 PVs become dilated in patients older than 50 years (Figure 1),3 which suggests mechanical effects of aging on AF triggers and substrates.3 Clinical data indicate that PV size correlates with PV arrhythmogenesis. Therefore, larger PVs may correlate with a higher PV arrhythmogenesis resulting in AF.

Aging dilates the diameter of the pulmonary vein (PV) orifices. Intra-atrial oblique-sagittal CT views show an older patient (age 70–79 years, C) with 4 dilated PVs compared with younger patients: 50–59 years, B; >40 years, A). (D) Average data for the left atrial appendage (LAA), left superior PV (LSPV) and left inferior PV (LIPV) trunk diameters in patients of different ages. *P<0.05. (Modified with permission from Pan NH, et al.3)
Both animal and human studies had shown that loss of cardiomyocytes by apoptosis and necrosis produces compensatory cellular hypertrophy, which may represent the structural basis of the aged heart, and contribute to cardiac dysfunction in the elderly.5 Aging increases the rate of ventricular cardiomyocyte death caused by enhanced vulnerability to stress such as oxidative stress.6 These histopathological changes at the microscopic level potentially induce AF occurrence or persistence.
Microcirculatory Disorder Defects in the coronary vasculature and microvasculature commonly occur in aging and contribute to myocyte cell death. Although microcirculation dysfunction in the atrium is still controversial in AF,7 capillary rarefaction has been frequently found in the LA posterior wall of AF patients.8 Additionally, chronic AF patients have a lower LA capillary density.9 Accordingly, aging-related microcirculatory dysfunction may play a role in the pathophysiology of AF.
Myocardial Fibrosis Age positively correlates with the severity of atrial fibrosis and apoptosis,5 which provides the structural support facilitating the occurrence of AF through greater conduction disturbances. However, the relationship between aging and atrial fibrosis is still controversial. Different from previous pathological studies of the human atrial appendage in open heart surgery, Platonov et al reported no correlation between age (<60 vs. >60 years old) and fibrosis in human autopsy studies.10 These discrepancies in the effects of aging on atrial fibrosis might be caused by different study populations, limited age-related increase in fibrosis extent, or heterogeneous atrial interstitial fibrosis. Cell loss with fibrosis replacement is commonly found in the aged LA (Figure 2).11 Old rat atrium shows a significant increase in highly heterogeneous interstitial fibrosis.12 This nonuniform atrial interstitial fibrosis could promote conduction slowing, wavefront breakups, and AF maintenance in aging. Fibrosis has significant arrhythmogenetic effects on PV cardiomyocytes. Collagen, the major element of fibrosis, directly increases spontaneous activity in PV cardiomyocytes through p38 mitogen-activated protein kinase activation.13 Moreover, direct incubation of collagen may lead to a shorter action potential duration (APD) in atrial myocytes.13 However, the previous human studies showed that the atrial effective refractory period (ERP) was unchanged or even prolonged with advancing age.14–16 This inconsistency suggests that collagen-induced APD shorting may be localized and heterogeneous, which increases focal atrial dispersions of refractoriness and facilities the genesis of micro-reentry. Collagen can produce larger Ca2+ transients and a larger calcium content in the sarcoendoplasmic reticulum (SR) with higher expression of SERCA2a, which is caused by downregulation of Thr17-phosphorylated phospholamban in collagen-treated atrial myocytes.17 These findings suggest that intracellular Ca2+ overload may contribute to aging-related atrial tachyarrhythmia.

Histological analysis (Masson trichrome staining) of an adult and aged left atrium (LA) of a rabbit. There are areas of cell loss with marked fibrosis, and hypertrophy of cardiomyocytes characterized by large-volume nuclei and increased fiber diameter in the aged rabbit LA (A), as compared with normal cardiomyocytes in the adult rabbit LA (B).
Myocyte Modification Aging-associated cardiomyocyte modification includes senescence, apoptosis or autophagy in response to accumulation of stress.18 To compensate for the cell loss, atrial myocytes develop hypertrophy as an adaptive mechanism. However, cellular hypertrophy may be insufficient to meet the functional demands caused by continuous loss of myocytes in advanced age, which eventually leads to atrial dysfunction.
Cellular Hypertrophy Human study has shown that compensatory enlargement of remaining myocytes for the loss of myocytes is frequently found in the aged ventricles.19 In a rat model hypertrophic LA myocytes had a high arrhythmogenic potential with greater incidence and frequency of spontaneous Ca2+-transients.20
Connexin Modification Gap junctions are composed of narrow membrane channels that permit small molecules to diffuse intercellularly. In rat ventricular cardiomyocytes, gap junctions provide low-resistance electrical coupling between adjacent cardiac myocytes.21 Aged rat myocardium has disruptions in the distribution of connexin (Cx)43, which may provide a cellular basis for aging-related cardiac arrhythmia.21 Loss of the gap junction may contribute to the electromechanical dysfunction associated with hibernation or predispose the heart to reentry arrhythmias in infarct border zones.22 Gap junctions impaired by heptanol have reduced greater electrical activity in the sinoatrial node (SAN) compared with PVs of rabbits, which may facilitate ectopic electrical activity and enhance the occurrence of AF.23 In contrast, aging is associated with an increase in Cx40 expression and a corresponding increase in gap-junctional resistivity. A canine study revealed that the gap junction-Cx43 distribution in the free wall of the right atrium (RA) with increasing age was more polarized and localized to the cell, causing lateral uncoupling, which was further enhanced by widening of interstitial spaces.24 That study also found that an elliptical activation pattern changed with aging to a square activation pattern with decreased incidence of longitudinal block but increased incidence of transverse block. Taken together, aging leads to different curvature of wave fronts and preferential direction of conduction block, which might play a role in multiple reentry and predispose to a higher incidence of AF.
Aging carries a lot of AF risk factors including heart failure (HF), endothelial dysfunction, etc. These indirect aging effects may increase AF occurrence through structural and electrophysiological remodeling.
Aging-Related Disorder: HFAging is also associated with an increased prevalence of HF, which is characterized by chamber dilatation including the LA and PVs. HF may increase the occurrence of AF through substrate modulation. Moreover, in an animal study HF epicardial fat possibly enhanced atrial arrhythmogenesis through modulation of the RA and LA electrophysiological properties.25 The elevated LA pressure secondary to increased LA stiffness in the setting of diffuse LA fibrosis might be associated with AF recurrence.26 Therefore, aging-induced structural changes contribute significantly to the pathophysiology of AF.
Aging-Related Disorder: Endothelial DysfunctionThe vascular endothelial dysfunction that occurs with aging is associated with a progressive reduction in the bioavailability of nitric oxide (NO) and an increase in the production of cyclooxygenase (COX)-derived vasoconstrictor factors.27 NO has been shown to rescue Ca2+ overload-induced triggered arrhythmias in ventricular myocytes.28 An NO donor (nitroprusside) was shown to decrease PV cardiomyocytes’ spontaneous beating rates, shorten the APD, and suppress delayed afterdepolarizations in a rabbit model.29 An NO donor also could inhibit L-type calcium currents (ICa-L), transient outward currents (Ito), and transient inward currents, but increase delayed rectified potassium currents. NO and NO donors inhibit Ito and the ultrarapid delayed rectifier channel recorded in human atrial myocytes.30 Still, these findings indicate that NO can regulate PV arrhythmogenesis by modulating calcium handling and other ionic currents through mechano-electrical feedback.
The shortening of the APD and the atrial ERP will facilitate the genesis of reentrant circuits based on the wave length theory, whereas shortening of the APD is a hallmark of “AF begets AF”. In addition, dispersion of the ERP because of regional difference in aging-related action potential characteristics might provide the basis for reentry, further leading to the occurrence or maintenance of AF. Trigger activity plays a critical role in the pathophysiology of paroxysmal AF (PAF), as increasing calcium leakage with dysregulated calcium protein has been demonstrated in PAF.31 Aging may regulate atrial and PV electrical activity through mechano-electrical feedback, calcium dysregulation, oxidative stress, and metabolic disorders.32–34
Aging Effects on Electrophysiological Characteristics and Ionic Currents There have been many studies investigating aging-related changes in the ERP and they are controversial.12,14–16,35 Some human studies revealed significant correlations between age and atrial ERP and a progressive increment of dispersion of atrial refractoriness,15 while others reported no age-related increase in atrial dispersion.35 Unchanged ERP was reported in some human studies, but these were done performed at a single RA site and/or with 1 cycle length, thus are limited by these restrictions.16 Similarly, the duration of the monophasic action potential shows no progressive trend with age.36 Moreover, an animal study found that the APD90 and ERP are prolonged in the aged RA, but shortened in the aged LA.37 The discrepancies between the action potentials in the RA and LA and the shortening of the atrial APD may provide a substrate for reentry and tendency to AF maintenance.
A canine study revealed that the INa density in the LA was greater than in the RA in both adult and aged atrial myocytes, but the difference in INa density between atrial chambers was unaffected by age.38 The similar expression of Nav1.5 in the adult and aged RA or LA suggests that the cardiac Na+ channel protein (Nav1.5) is not significantly affected by aging.38 Aging has been shown to increase INa-Late in ventricular myocytes.39 Aging-related disorders such as HF also increase INa-Late in PV cardiomyocytes.40 However, the effects of aging on PV or atrial INa-Late have not been fully elucidated.
The densities of the Ito and Isus (sustained potassium current) are significantly increased in the aged canine RA,41 but this is not observed in aged LA cells.42 The aging-related increase in Isus together with a decrease in ICa-L may result in either an unchanged or prolonged ERP in the RA. The acetylcholine-induced potassium current (IKACh) plays a vital role in vagal-induced AF and increases the risk of AF in aging.42
Aging Effects on Calcium Dysregulation A study of guinea pigs found that expression of the Cav1.2 protein within the sinoatrial node (SAN) declines during aging accompanied by a reduction in SAN electrical activity.43 Aged PVs of rabbits have larger amplitudes of delayed afterdepolarization (DAD), greater depolarized resting membrane potential (RMP), longer APDs, and higher incidence of both AP alternans and contractile alternans, which suggests abnormal calcium regulation may underlie increasing arrhythmogenesis in aging PVs.44 Aged PVs are more susceptible to SR Ca2+ leakage caused by increased ryanodine receptor (RyR) (Figure 3),44 which supports the concept that RyR dysfunction may result in high PV arrhythmogenesis and aging-related arrhythmogenic vulnerability.

Aging increases the arrhythmogenesis of rabbit pulmonary veins (PV). The tracings (A) and average data (B) show that rapamycin 1 nmol/L increased the beating rate in aged PVs but not in young PVs. (C) Rapamycin (1 nmol/L)-induced burst firing in aged PVs. (Modified with permission from Wongcharoen W, et al.44)
Aged rabbit LAs tend to have a higher incidence of DAD (Figure 4).33 Moreover, a study of isolated human RA myocytes revealed that aging was associated with depression of the SR calcium content and calcium transient amplitude, which may favor a progressive decline in RA contractile function with age.34
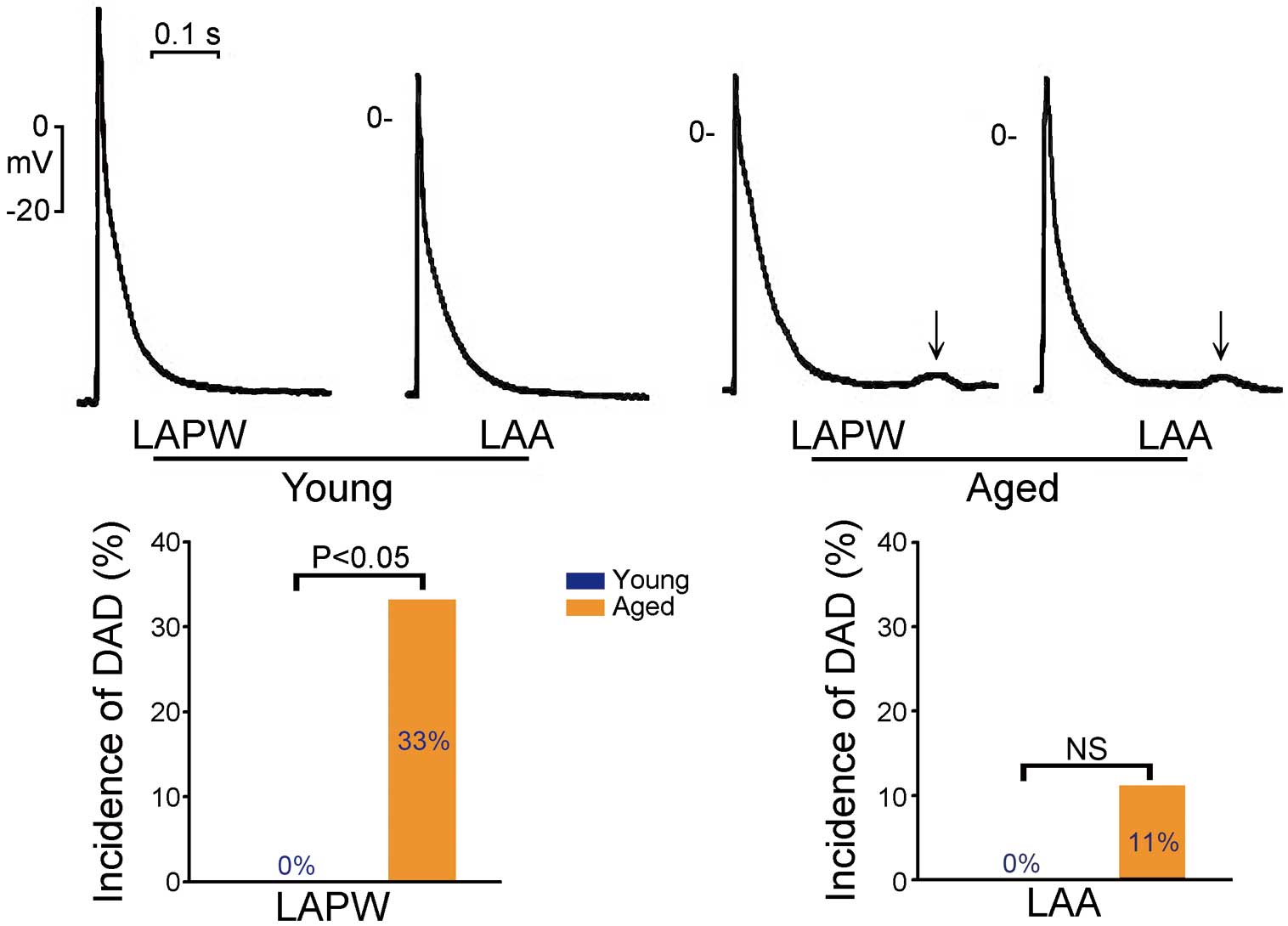
Aging increases triggered activity in the rabbit atrium. (Upper panels) Larger delayed afterdepolarization (DAD) (↓) in aged rabbit left atrial posterior walls (LAPWs) and left atrial appendages (LAAs). (Lower panels) Higher incidence of DAD in aged LAPWs than in young LAPWs, but not in LAAs. NS, non-significant difference. (Modified with permission from Wongcharoen W, et al.33)
HF causes significant atrial electrical remodeling with a calcium overload because of increasing SERCA and calcium leakage Theoretically, aging is expected to dysregulate calcium homeostasis and lead to atrial arrhythmogenesis. Similarly, rabbit HF PV cardiomyocytes had faster spontaneous activity and a higher incidence of DAD.40 Moreover, rabbit HF PV cardiomyocytes have more Ca2+ sparks. Therefore, aging may dysregulate sodium and calcium homeostasis in PV cardiomyocytes in conditions such as HF.
Aging Effects on Mechano-Electrical Feedback Mechano-electric feedback refers to the phenomenon of mechanical load on myocardial tissue modulating the electrical function of the heart by altering the electrical function of the cells, which can change APD or generate extra beats.45 Stretch induces membrane depolarization, prolongation of the AP and afterdepolarizations in isolated cardiomyocytes.45
Aging-associated cardiac dysfunction is a condition of altered mechanical loading that may potentiate mechano-electrical feedback. Mechano-electrical feedback is the critical link in structural and electrical remodeling.46 A dilated atrium or PV not only provides substrate support for reentry, but also significantly changes the atrial (or PV) electrical properties.2,31,47 An animal study revealed that stretch force dependently increased the incidence of spontaneous activity, early afterdepolarization (EAD) and DAD in the PVs.46 Moreover, stretch-activated channels blockers, gadolinium and streptomycin, decreased the incidence of spontaneous activity and firing rates in PVs. These findings indicate that stretch-induced arrhythmogenic activity of the PVs may contribute to the genesis of AF.
Racial differences exist in AF occurrence. Whites have higher rates of AF than Blacks, despite a lower prevalence of traditional AF risk factors.48 Clinical study suggested that a higher AF rate in Caucasians than in African-Americans might be related to a larger LA diameter.49 Regardless of the elevated AF risk from aging, African-Americans had a 41% lower age- and sex-adjusted risk of being diagnosed with AF compared with Whites.50 Moreover, another clinical study revealed that systemic inflammatory biomarkers such as adiponectin, tumor necrosis factor (TNF)-α, TNF-α SR I, and TNF-α SR II concentrations were each higher among Whites and independently associated with a greater risk of incident AF,51 which suggests systemic inflammatory pathways significantly mediate the heightened risk of AF among Whites.
Aging is associated with a decline in sex hormones and increased incidence of AF in both men and women. Human studies of the left and right ventricles have revealed that the aging process leads to myocyte cell loss and myocyte cellular reactive hypertrophy in men but not in women, suggesting a sex difference in the aging heart.52 Male sex carried a 1.5-fold risk of developing AF after adjusting for age and other predisposing conditions. However, menopause did not increase the incidence of AF in women but androgen deprivation therapy increased it in men.53 Testosterone deficiency in the elderly strongly increases the risk of AF; the Framingham heart study showed that in men aged ≥80 years, a 1 standard deviation decrease in testosterone was associated with a hazard ratio of 3.53.54 In rat atria, deficiency of testosterone was arrhythmogenic, with less binding of FKBP12.6 to RyR2 possibly inducing calcium leakage from the SR.55 Accordingly, testosterone deficiency may contribute to the risk of AF in aging males. However, animal experiments showed that testosterone replacement increases arrhythmogenesis in aged PVs and LA, but testosterone-treated PVs had a faster spontaneous rate, larger amplitude of angiotensin II-induced DAD and higher incidence of isoproterenol-induced EAD and DAD.11,56 In addition, there is a high probability of H2O2-induced burst firing and EAD in testosterone-treated PVs, and more isoproterenol-induced DAD and spontaneous activity in the testosterone-treated LA.11 The arrhythmogenic effects of testosterone treatment in aging animals may be caused by the non-physiological replacement. It is possible that timely treatment of aging-related androgen deficiency with different formulations may lead to different outcomes.
Aging humans tend to have higher levels of acute-phase inflammatory cytokines, including C-reactive protein, interleukin-6, and TNF-α.57 Inflammation contributes to the pathophysiology of AF. Moreover, the proinflammatory cytokine, TNF-α, directly increases PV arrhythmogenicity with dysregulated calcium homeostasis.58 Therefore, the aged may have a propensity for a higher risk of inflammation-induced AF.59 Adipose tissues can secrete numerous hormones and cytokines that can induce both proinflammatory and protective factors.60
Reactive oxygen species are suggested to be one of the primary determinants of aging.61 Aging can increase oxidative stress; oxidative stress is related to mitochondrial function, and AF has been reported to be associated with accumulation of aging-related mitochondrial DNA (mtDNA) deletion mutations.62,63 Accumulation of 4977-base-pair mtDNA deletion mutations in the human RA appendage is associated with AF, similar to the aging process of atrial tissue, which might contribute to the atrial dysfunction in AF.64 Another study found that a mtDNA deletion of 7.4 kb was significantly associated with age, the prevalence of AF, and a decreased concentration of adenine nucleotides in the atrium, suggesting a bioenergetic deficiency caused by impaired ATP synthesis in the human atrium.63 Age-related histological changes (interstitial fibrosis and apoptosis) were suppressed by pioglitazone in a rat study, suggesting that peroxisome proliferator-activated receptor gamma activator may prevent age-related AF through inhibiting the mitochondrial apoptotic signaling pathway.64 H2O2 concentration-dependently induced the occurrence of burst firing in aged PVs with spontaneous activity.32,65 Oxidative stress may facilitate the occurrence of PV burst firing and the genesis of EAD with the known greater Ca2+ content in the SR, Ca2+ transients, and the less-negative RMP in PVs with spontaneous activity.32 Because both inflammation and oxidative stress influence anatomical as well as functional properties, they could mediate or bridge the interaction between structural or electrophysiological remodeling.
Metabolic disorders are common with aging, and include mitochondrial dysfunction, insulin resistance, and dysregulated intracellular lipid metabolism.66 Stress-related metabolic changes and altered activity of AMP-activated protein kinase (AMPK) are proposed to play an important role in the pathogenesis of AF67 because AF is characterized by irregular contractions of atrial cardiomyocytes and increased energy demands. Currently, laboratory evidence suggests a potential role of AMPK in the pathophysiology of AF.67 Irregular pacing of cardiomyocytes mimicking AF increases diastolic Ca2+ levels and upregulates Ca2+/calmodulin-dependent protein kinase II and AMPK, which in turn increases membrane expression of fatty acid translocase (FAT/CD36), resulting in lipid accumulation in the LA of patients with AF compared with matched samples of patients in sinus rhythm.68 These observations revealed the existence of ‘‘metabolic remodeling’’ in addition to structural and electrical remodeling, which may contribute to the pathology of AF.68 Moreover, cardiomyocyte-specific ablation of liver kinase B1, an activator of AMPK, in a mice model resulted in the development of spontaneous AF and an age-dependent progression of AF from a paroxysmal to a persistent form.69 Collectively, these findings again suggest the importance of atrial metabolic remodeling in AF.
Sick sinus syndrome (SSS) and AF both markedly increase in incidence and prevalence over time, and are common in aged populations.70 Low heart rates (HR) are associated with an increased risk of AF in elderly people.71 HR is reduced whereas SAN recovery time, a direct measure of SAN function, is prolonged in the aged human or animal.35,72 Animal studies reveal downregulation of hyperpolarization-activated cyclic nucleotide gated potassium channel 4 (HCN4), diminished expression of the Cav1.2 protein, and loss of Cx43 with increased distance travelled by the impulse generated at the center of the aged SAN to the atrial tissuen,73,74 which provides a molecular correlation to the gradually increased incidence of SAN dysfunction with aging. The combination of anatomical and electrophysiological remodeling lays the groundwork for clinical presentations of both SSS and AF. Moreover, SSS facilitates the development of AF through an increase in the likelihood of atrial ectopics caused by escape from the overdriving sinus node. Intact electrical connections between the SAN and PVs is crucial for preventing arrhythmogenesis, as demonstrated by experiments in which rabbits with disrupted electrical connections between the SAN and PVs demonstrated more burst firing and EAD in response to Anemonia sulcata toxin (ATX)-II (Figure 5).75 However, the reverse view is also supported by clinical observation that longstanding AF and other supraventricular tachycardias cause remodeling of the SAN, leading to its dysfunction and subsequent bradycardias, which might recover after ablation of the tachyarrhythmias.

Interactions of sinoatrial node (SAN) and pulmonary vein (PV) electrical activity determine the occurrence of PV arrhythmogenesis. Examples (Upper panel) and average data (Lower panel) of frequent occurrences of burst firing (*) in isolated PV preparations after disconnection. ATX, Anemonia sulcata toxin; SVC, superior vena cava. (Modified with permission from Chen YC, et al.75)
In conclusion, aging is associated with multiple anatomical, electrophysiological, ionic, and metabolic changes, either directly related to aging or to aging-related comorbidities and cardiovascular disorders, which all subsequently lead to the occurrence of AF.
This work was supported by grants from Taipei Medical University-Wan Fang Hospital (104swf02, 104-wf-eva-01, 105-wf-eva-08 and 105-wf-eva-14).
None declared.