Abstract
Background:
Bridge-to-decision (BTD) devices providing temporary mechanical circulatory support should be introduced to patients with advanced heart failure. This study evaluated the effectiveness and safety of a BTD device comprising an innovative extracorporeal continuous-flow temporary ventricular assist device (VAD) driven by a novel hydrodynamically levitated centrifugal flow blood pump.
Methods and Results:
Nine patients, comprising 3 with dilated cardiomyopathy, 3 with fulminant myocarditis, and 3 with ischemic heart disease, and 6 males, whose mean age was 47.7±8.1 years, were enrolled into the study. Six patients had Interagency Registry for Mechanically Assisted Circulatory Support profile 1, and 3 were profile 2. The primary endpoint was a composite of survival free from device-related serious adverse events and complications during circulatory support. Eight patients received left ventricular support, of whom 3 received concomitant right ventricular support using extracorporeal membrane oxygenation circuits, as a consequence of severe respiratory failure. One patient with fulminant myocarditis received biventricular support using the novel VAD system. After 19.0±13.5 days, 3 patients were weaned from circulatory support, because their native cardiac function recovered, and 6 patients required conversion to a durable device as a bridge-to-transplantation. One patient had non-disabling ischemic stroke episodes, and no patients died.
Conclusions:
This novel extracorporeal VAD system with a hydrodynamically levitated centrifugal pump can safely and successfully bridge patients with advanced heart failure to subsequent therapeutic stages.
Ventricular assist devices (VADs) have long been recognized as essential treatment options for patients with advanced heart failure who do not respond to conventional guideline-directed medical therapies.1
Using implantable continuous-flow (CF)-VADs in patients with advanced heart failure has totally changed the advanced heart failure care landscape. Patients supported by CF-VADs benefit greatly regarding their prognoses, and their quality of life improves considerably, enabling them to return to almost ordinary lives.2,3
In contrast, patients with acutely deteriorating heart failure, including cardiogenic shock, cannot receive durable implantable CF-VADs directly, but they can receive temporary mechanical circulatory support to stabilize them initially and provide bridges to the next therapeutic stages.4,5
To date, a variety of temporary mechanical circulatory devices have been used for these bridge-to-decision (BTD) strategies.6–8
Centrifugal flow pump-driven VADs are more durable and dependable, and are one of the more feasible BTD devices, and more thromboresistant high-flow centrifugal pumps are always required to provide better life-saving therapeutic options.9,10
However, no centrifugal flow pump-driven VAD systems for long-term use have not been reimbursed in our country so far.
Recently, we developed a new extracorporeal CF temporary VAD system (BR16010) that is driven by a novel disposable centrifugal flow blood pump with a hydrodynamically levitated bearing. This system includes a pump, cannulae, connectors, and circuits, and it has been shown to provide effective and safe support in a preclinical study involving a goat model.11,12
Following these promising preclinical results, we designed the first in-human clinical study (NCVC-BTD_01),13
and the purpose of the current study was to evaluate the safety and effectiveness of this system as a BTD therapy for patients with advanced heart failure and/or refractory cardiogenic shock.
Methods
Study Design and Ethical Considerations
NCVC-BTD_01 is being conducted at the National Cerebral and Cardiovascular Center in Japan, and it is a single-center, single-arm, open-label, exploratory, investigator-initiated, first in-human clinical study of a medical device. The study complied with the principles of the Declaration of Helsinki, and it was conducted in accordance with the International Conference on Harmonization/Good Clinical Practice guidelines, the International Organization for Standardization 14155:2011: Clinical investigation of medical devices for human subjects—Good clinical practice, and country-specific requirements. The study was reviewed and approved by the National Cerebral and Cardiovascular Center’s Institutional Review Board on 30 July 2017. The study was registered in the UMIN Clinical Trials Registry (UMIN-CTR: R000033243). Written informed consent was obtained from the enrolled patients or their families at study enrollment.
Study Subjects and Sample Size
The study subjects comprised patients with advanced heart failure or refractory cardiogenic shock who were unresponsive to conventional medical and/or surgical treatments or mechanical circulatory support, including intra-aortic balloon pumps (IABPs), venoarterial extracorporeal membrane oxygenation (ECMO), percutaneous cardiopulmonary support (PCPS), and other VAD systems, and were eligible for rescue therapy using VADs as BTD or bridge-to-recovery strategies. Detailed information of inclusion criteria and exclusion criteria of this study were previously described (Supplementary Table 1).13
A total of 9 patients were enrolled in the study. The study was planned using Simon’s minimax 2-stage design, as described previously.13,14
Briefly, the hypothesis to be tested was H0: P<P0 vs. H1: P>P1, where P was the probability of mortality at 30 days, a false-positive rate (α) ≤10% and a false-negative rate (ß) ≤10% were accepted, P0 was set at 50%, and P1 was set at 90%. Within these constraints, 4 patients were enrolled for the first stage. The study would stop if ≤2 patients survived, would continue until a total of 9 patients were accrued, and be declared a success if >7 patients survived.
Endpoints
The study’s primary endpoint was a composite of survival free from device-related serious adverse events and complications during device support. Regarding left ventricular (LV) assistance, withdrawal of the BTD device as a consequence of native LV functional recovery or exchange to a durable implantable VAD as a bridge-to-transplantation within 30 days after implantation was considered a study success. Regarding right ventricular (RV) assistance, BTD device withdrawal as a consequence of native RV functional recovery within 30 days after implantation was considered a study success.
The secondary endpoints included the VAD support period, and changes in the brain natriuretic peptide (BNP) levels, LV ejection fraction (LVEF), and LV diastolic dimension (LVDd) at 7 days after device implantation and on the day of device withdrawal. To evaluate the effect of the device on end-organ function, changes in the creatinine (Cre), total bilirubin (T-Bil), and aspartate aminotransferase (AST) levels were assessed before and after VAD implantation. Changes in lactate acid dehydrogenase (LDH) and indirect bilirubin (I-Bil) during the device support period were also assessed to determine the occurrence of hemolysis.
BTD Device
The BTD device has been described in detail previously.13
Briefly, the VAD system comprises a disposable centrifugal flow pump with a hydrodynamically levitated bearing (BR16010), cannulae, an extracorporeal circuit, and metallic connectors (Figure 1). Flexible polyvinyl chloride (PVC) tubes (1/2 inch diameter) with hard tips are used for the inflow cannula (Figure 1A), a 12-mm diameter vascular prosthesis attached to a flexible PVC tube is used for the outflow cannula (Figure 1B), and custom-made metallic connectors are used to connect the cannulae and the extracorporeal circuit (Figure 1C). The extracorporeal circuit consists of a PVC tube (70 cm long and 3/8 inch wide) coated with T-NCVC®
heparin (Toyobo, Osaka, Japan) (Figure 1D). The pump’s priming volume is 18 mL (Figure 1E). The impeller is embedded in a stationary casing and it rotates with a narrow clearance at both ends (Figure 1F). The blood film in this space serves as the journal bearing, which generates sufficient pressure to sustain the impeller and avoid mechanical contact with the stationary parts. The console weighs ~8 kg, is 210 mm wide, 350 mm deep, and 326 mm high, and is usually mounted on a frame with wheels, an adaptor for the pump, and a handle to reposition the device (Figure 1G). The clinical parameters, including the pump’s rotational speed and flow, are displayed on a screen on the console, and the pump’s rotational speed can be adjusted easily at the console.
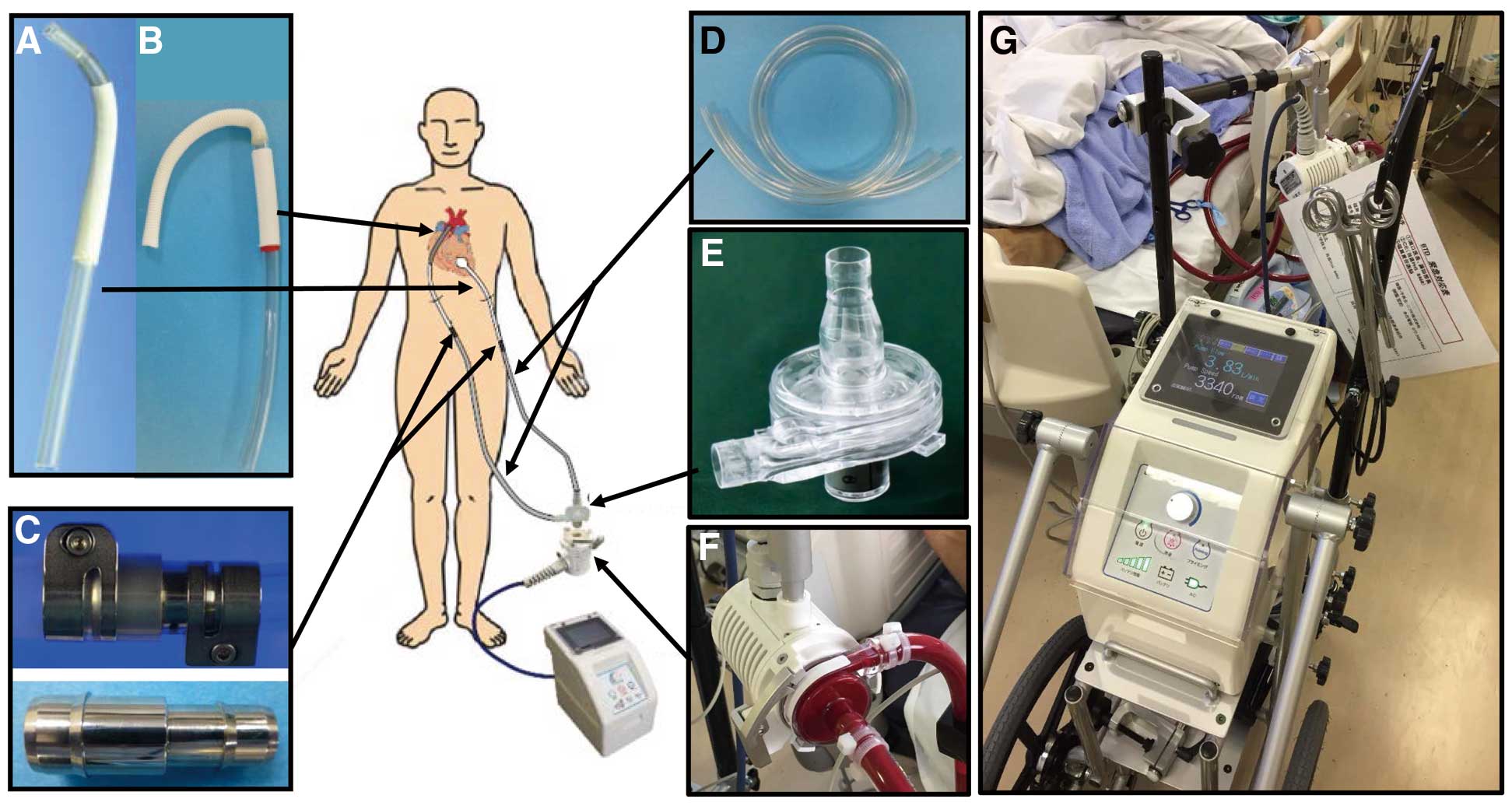
All of the surgical procedures were performed through a median sternotomy under a cardiopulmonary bypass. To implant the left VAD (LVAD), the outflow cannula was anastomosed to the ascending aorta, and the inflow cannula was anastomosed to the LV apex. The extracorporeal circuit was then connected to the pump and placed between the inflow and outflow cannula. To implant the right VAD (RVAD), the inflow cannula was placed at the RV apex, and the outflow cannula was anastomosed to the main pulmonary artery. If there was no surgical bleeding, heparin infusions began 24 h after surgery to maintain the activated partial thromboplastin time within 50–60 s. Aspirin (100 mg daily) and warfarin were introduced 24 h after surgery, and the warfarin dose was adjusted to maintain a target international normalized ratio of 3. The heparin infusion was then replaced by oral aspirin and warfarin.
Statistical Analyses
The continuous variables are expressed as the means and the standard deviations (SDs) or as the medians and the interquartile ranges (IQRs), as appropriate. The categorical variables are expressed as counts and frequencies. Comparisons between groups were performed using unpaired t-tests for normally distributed data, or the Mann-Whitney U-test for non-normally distributed data. The statistical analyses were performed using JMP software, version 13 (SAS Institute Inc., Cary, NC, USA).
Results
Four patients were enrolled to participate in the first stage of the study in accordance with the 2-stage design. As all of these patients survived >30 days, 5 additional patients were enrolled to participate in the second stage of the study. The first patient was enrolled on 6 October 2017, and the last patient was enrolled on 8 March 2018.
Table 1
presents the patients’ baseline demographic and clinical data. All of the patients were Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) profiles 1 and 2 at enrollment, and 8 patients had received temporary circulatory support, including IABPs and/or PCPS, at study enrollment. All laboratory data were obtained while patients were on temporary mechanical circulatory support.
Figure 2
provides an overview of the statuses of the study subjects at the time of enrollment, the devices used, and the outcomes.
Table 2
provides detailed data describing the clinical parameters and each enrolled patient’s course, including their therapeutic strategies and outcomes. Five patients received LV support and 1 patient received biventricular support using the BTD device. The 3 remaining patients who had critical respiratory dysfunction caused by severe pulmonary congestion received LV support using the BTD device concomitant with RV support using an RVAD and a conventional ECMO circuit (RVAD-ECMO). Five patients with LVADs and 1 patient with a LVAD concomitant with a RVAD-ECMO could not be weaned from mechanical circulatory support, and they received an implantable durable LVAD as a bridge-to-transplantation. Two patients with LVADs concomitant with RVAD-ECMOs and 1 patient with a biventricular support assist device were successfully weaned from the VADs, because their native cardiac function recovered. The mean VAD support period was 19.0±13.5 days (range, 9–48 days), and during the study period that included 7 days after device removal, 1 patient experienced 2 non-disabling ischemic stroke episodes, and none of the patients died. Regarding other device- or surgery-related complications, 2 patients developed cardiac tamponade that required chest reopening, and a cannula injury was detected in 1 patient that probably occurred when the cannula and circulatory circuit were being connected using the metallic connectors. With respect to hemolysis, values of LDH and I-Bil during the support period are shown in
Supplementary Table 2. Although there was elevation in LDH in some cases early after conversion or implantation of a VAD, the values decreased with time, and there are no clinically meaningful elevations suggesting occurrence of hemolysis.
Table 1.
Patients’ Baseline Demographic and Clinical Data
| |
Age
(years) |
Sex |
Etiology |
Height
(cm) |
Weight
(kg) |
BSA
(m2) |
INTERMACS
profile |
Preoperative MCS |
LVDd
(mm) |
LVEF
(%) |
T-Bil
(mg/dL) |
Cre
(mg/dL) |
BNP
(pg/mL) |
| Devices |
Duration
(days) |
| Case 1 |
56 |
M |
AMI |
170 |
60 |
1.69 |
1 (TCS) |
C-ECMO |
7 |
42 |
20 |
4.3 |
0.77 |
773.8 |
| Case 2 |
54 |
F |
DCM |
158 |
56.4 |
1.57 |
2 |
None |
11 |
64 |
10 |
2.5 |
0.5 |
234.8 |
| Case 3 |
50 |
M |
OMI |
184 |
86.9 |
2.1 |
2 (TCS) |
P-LVAD |
24 |
70 |
7 |
1.6 |
0.55 |
663 |
| Case 4 |
48 |
F |
DCM |
160 |
40 |
1.37 |
2 (TCS) |
IABP |
1 |
66 |
20 |
0.7 |
0.28 |
1,753.8 |
| Case 5 |
49 |
M |
AMI |
182 |
80 |
2.01 |
1 (TCS) |
LVAD-ECMO |
18 |
52 |
10 |
3.4 |
1.1 |
575 |
| Case 6 |
45 |
M |
Myocarditis |
164 |
66 |
1.72 |
1 (TCS) |
P-ECMO |
1 |
47 |
8 |
0.7 |
0.77 |
392.6 |
| Case 7 |
42 |
M |
Myocarditis |
178 |
70 |
1.87 |
1 (TCS) |
P-ECMO |
2 |
52 |
5 |
1.2 |
3.94 |
226 |
| Case 8 |
55 |
M |
DCM |
175 |
80 |
1.96 |
1 (TCS) |
P-ECMO |
1 |
62 |
5 |
1.7 |
1.57 |
471.9 |
| Case 9 |
30 |
F |
Myocarditis |
146 |
40 |
1.28 |
1 (TCS) |
IABP |
1 |
38 |
5 |
1 |
0.59 |
547.1 |
AMI, acute myocardial infarction; BNP, brain natriuretic peptide; BSA, body surface area; C-ECMO, central extracorporeal membrane oxygenation; CHDF, continuous hemodiafiltration; Cre, creatinine; DCM, dilated cardiomyopathy; F, female; IABP, intra-aortic balloon pumps; INTERMACS, interagency registry for mechanically circulatory support; LVDd, left ventricular diastolic dimension; LVEF, left ventricular ejection fraction; M, male; MCS, mechanical circulatory support; OMI, old myocardial infarction; P-ECMO, peripheral extracorporeal membrane oxygenation; P-LVAD, paracorporeal left ventricular assist device; T-Bil, total bilirubin; TCS, temporary circulatory support.

Table 2.
Patients’ Clinical Parameters and Each Enrolled Patient’s Course, Including Therapeutic Strategies and Outcomes
| |
Operative
time
(min) |
Bypass
time
(min) |
Initial support
strategies |
Support
time
(days) |
Pump
speed
(rpm) |
Pump
flow
(L/min) |
PCWP
(mmHg) |
PAP
(mmHg) |
RAP
(mmHg) |
CI
(L/min−1/m−2) |
Adverse
events |
Outcome |
| Case 1 |
251 |
102 |
LVAD |
21 |
3,400 |
4.15 |
4 |
23/9 (13) |
4 |
2.54 |
Cerebral
infarction |
BTB |
| Case 2 |
274 |
142 |
LVAD |
11 |
3,000 |
3.67 |
– |
30/16 (22) |
10 |
2.7 |
Cannula
injury |
BTB |
| Case 3 |
– |
– |
LVAD |
10 |
4,000 |
5.24 |
– |
28/12 (18) |
8 |
2.7 |
None |
BTB |
| Case 4 |
164 |
52 |
LVAD |
13 |
3,030 |
3.51 |
9 |
30/15 (20) |
8 |
2.8 |
None |
BTB |
| Case 5 |
89 |
– |
LVAD |
34 |
4,810 |
5.18 |
– |
18/11 (15) |
9 |
2.5 |
None |
BTB |
| Case 6 |
251 |
110 |
BiVAD
(BR16010) |
79 |
L: 3,900
R: 3,300 |
5.00
3.35 |
– |
20/11 (14) |
8 |
3 |
None |
BTR |
| Case 7 |
174 |
63 |
LVAD (BR16010)
+RVAD-ECMO |
9 |
3,210 |
4.20 |
– |
16/13 (14) |
5 |
2.1 |
Symptomatic
epilepsy |
BTR |
| Case 8 |
288 |
100 |
LVAD (BR16010)
+RVAD-ECMO |
16 |
3,330 |
3.45 |
– |
21/14 (16) |
7 |
2.1 |
Cardiac
tamponade |
BTR |
| Case 9 |
211 |
78 |
LVAD (BR16010)
+RVAD-ECMO |
48 |
3,020 |
3.52 |
6 |
18/12 (14) |
12 |
2.87 |
Cardiac
tamponade |
BTB |
BiVAD, biventricular assist device; BTB, bridge-to-bridge; BTR, bridge-to-recovery; CI, cardiac index; ECMO, extracorporeal membrane oxygenation; PAP, pulmonary arterial pressure; PCWP, pulmonary capillary wedge pressure; LVAD, left ventricular assist device; RAP, right atrial pressure; RVAD, right ventricular assist device.
Figure 3
presents the changes in the BNP levels, LVEF, and LVDd. The median BNP level decreased significantly from 547.1 (IQR, 309–875) pg/mL at study enrollment to 278.7 (IQR, 185–391) pg/mL at VAD removal. The mean LVDd decreased significantly from 54.6±11.0 mm at study enrollment to 44.9±11.1 mm at 7 days post-VAD implantation, with no further decreases at VAD removal. The median LVEF increased significantly from 8% (IQR, 5–15%) at study enrollment to 15% (IQR, 15–37.5%) at VAD removal. The median Cre level, which was elevated at study enrollment as a consequence of systemic circulatory failure, decreased from 0.91 (IQR, 0.68–1.39) mg/dL to 0.49 (IQR, 0.39–0.71) mg/dL at 7 days post-VAD implantation (P=0.007) (Figure 4). The median AST level declined from 182 (IQR, 56–1,427) U/L at study enrollment to 36 (IQR, 26.5–57.5) U/L post-VAD implantation (P=0.07) (Figure 4). The T-Bil level, which increased significantly at 1 day post-VAD implantation, gradually decreased to baseline levels at 7 days post-VAD implantation (Figure 4).

Discussion
BTD therapy using our novel extracorporeal CF temporary VAD system and newly developed centrifugal flow blood pump, is a promising approach for patients with acutely deteriorating advanced heart failure. All of the enrolled patients with INTERMACS profiles 1 or 2 were successfully bridged to subsequent therapeutic stages, which included weaning them from the VADs as a consequence of native cardiac recovery and conversion to a durable implantable LVAD as a bridge-to-transplantation. Non-disabling strokes occurred in 1 patient; however, this patient was successfully bridged to a durable LVAD after 21 days of support. No other major critical adverse events occurred during support for a total of 171 days (mean, 19.0±13.5 days), except for cardiac tamponade.
VAD therapy for patients with critical conditions is always challenging. The prognoses for patients with lower INTERMACS profiles, for example, profile 1, are worse than those for patients with higher INTERMACS profiles after VAD implantation.15
Rescuing patients with critical conditions by using secure and safe surgical procedures and providing robust hemodynamic support with fewer device-related complications is mandatory. Hence, the extracorporeal CF temporary VAD system described in this study may be an appropriate BTD device. Regarding the system’s hemodynamic support capacity, despite the pump having a low priming volume, it can provide a flow rate of 5.0 L/min against a pressure head of 400 mmHg at 5,150 rotations per min (rpm),11
and the current VAD system provided sufficient hemodynamic support to a goat model.12
In the current clinical setting, our data additionally demonstrated that the VAD system provided full hemodynamic support, even for participants with body surface areas >2.0. Especially in case 3, conversion from NIPRO VAD to the current device immediately resulted in improvement of pulmonary congestion and peripheral organ dysfunction under 2.7 L/min−1/m−2
of cardiac index in BSA >2.0 m2, which suggested the superiority of the current VAD system compared with conventional NIPRO VAD in terms of the pump flow volume. Regarding the thrombogenicity of this device, no major thromboembolic events were observed during the preclinical trial involving the goat model, except for microscopic renal infarction. The data from this study demonstrated that minor, non-disabling stroke episodes occurred in 1 patient. Anticoagulation therapy was administered, which comprised heparin infusions during the acute phase after surgery, followed by oral aspirin and warfarin. As cardiac tamponade developed in 2 patients, milder anticoagulation therapy may be possible with this device, especially during the acute phase after implantation.
In addition to the system’s hemodynamic reliability and lower thrombogenicity, the small console is an important practical feature of this device. For patient transportation, the frame has wheels and a small turning circle, and the console itself, which weighs only 8 kg, is easily detached from the frame and placed on a bed or stretcher. The internal battery, which remains charged for ~60 min, should contain sufficient energy to enable patient transportation in different clinical settings. The portability of this device is also another clinical superiority compared to other conventional VAD systems such as NIPRO VAD.
The device’s lowest rotational speed of 3,000 rpm caused some inconvenience when we performed the LVAD off/weaning tests, because the device can provide almost full circulatory support at this rotational speed, depending on the patient’s size, and it is difficult to evaluate native cardiac function during these tests. However, during LVAD off/weaning tests, we clamp the outflow side of the circuit to restrict the blood flow volume, and because the blood flow volume is clearly indicated on the console’s screen, fine adjustments can be made during these tests.
This study’s design is an important aspect of this investigation. Simon’s minimax 2-stage design, which was adapted for this study, is often applied to phase II clinical studies that evaluate the safety and efficacy of targeted treatments in single-arm trials. The study’s design involved separating the patients into 2 groups or stages, and as the efficacy of the targeted treatment was greater than the predetermined threshold after the completion of the first stage, the second stage proceeded. In the current study, 4 patients were enrolled to participate in the first stage, followed by 5 patients in the second stage. Because the 2-stage design is usually applied to clinical trials of cancer treatments, we propose that this design could be applied to evaluate the efficacy of medical devices in critically ill patients.
The study’s limitations include its implementation at a single center, and the relatively small sample size. We calculated the number of study subjects required based on Simon’s minimax 2-stage design, as described in the methods section. On the basis of the current study’s findings, large scale, multicenter, prospective post-marketing surveillance studies, not only for LVAD use, but also for RVAD use, are recommended.
In conclusion, BTD strategies that involved a novel extracorporeal CF ventricular assist system and a hydrodynamically levitated centrifugal pump (BR16010), safely and successfully bridged patients with advanced heart failure to their next therapeutic stages. The current device can be used as a LVAD and a RVAD, which allows it to be used on patients with different types of advanced heart failure. We plan to use this device as a portable ECMO system in the future.
Acknowledgments
The authors thank Hideki Yotsuida, Yuzo Takahashi, Hiroshi Nishioka, Megumi Komiyama, Yuto Kimura, Maiko Hattori, Eri Miyoshi, Nobuaki Konishi, and Yumiko Hori for providing patient care.
Disclosures
None of the authors have any conflicts of interest to declare. This study was an investigator-initiated clinical study that was funded by research commission fees from Nipro Corporation, Osaka, Japan.
IRB Information
National Cerebral and Cardiovascular Center Institutional Review Board; Reference Number: NCVC-BTD_01.
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-19-1122
References
- 1.
Rose EA, Gelijns AC, Moskowitz AJ, Heitjan DF, Stevenson LW, Dembitsky W, et al. Long-term use of a left ventricular assist device for end-stage heart failure. N Engl J Med 2001; 345: 1435–1443.
- 2.
Miller LW, Pagani FD, Russell SD, John R, Boyle AJ, Aaronson KD, et al. Use of a continuous-flow device in patients awaiting heart transplantation. N Engl J Med 2007; 357: 885–896.
- 3.
Slaughter MS, Rogers JG, Milano CA, Russell SD, Conte JV, Feldman D, et al. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009; 361: 2241–2251.
- 4.
Riebandt J, Haberl T, Mahr S, Laufer G, Rajek A, Steinlechner B, et al. Preoperative patient optimization using extracorporeal life support improves outcomes of INTERMACS Level I patients receiving a permanent ventricular assist device. Eur J Cardiothorac Surg 2014; 46: 486–492; discussion 492.
- 5.
Loforte A, Marinelli G, Musumeci F, Folesani G, Pilato E, Martin Suarez S, et al. Extracorporeal membrane oxygenation support in refractory cardiogenic shock: Treatment strategies and analysis of risk factors. Artif Organs 2014; 38: E129–E141.
- 6.
Seguchi O, Fujita T, Watanabe T, Kuroda K, Hisamatsu E, Nakajima S, et al. Temporary biventricular support with extracorporeal membrane oxygenation: A feasible therapeutic approach for cardiogenic shock with multiple organ failure. J Artif Organs 2017; 20: 206–214.
- 7.
Kurihara C, Kawabori M, Sugiura T, Critsinelis AC, Wang S, Cohn WE, et al. Bridging to a long-term ventricular assist device with short-term mechanical circulatory support. Artif Organs 2018; 42: 589–596.
- 8.
den Uil CA, Akin S, Jewbali LS, Dos Reis Miranda D, Brugts JJ, Constantinescu AA, et al. Short-term mechanical circulatory support as a bridge to durable left ventricular assist device implantation in refractory cardiogenic shock: A systematic review and meta-analysis. Eur J Cardiothorac Surg 2017; 52: 14–25.
- 9.
Worku B, Pak SW, van Patten D, Housman B, Uriel N, Colombo P, et al. The CentriMag ventricular assist device in acute heart failure refractory to medical management. J Heart Lung Transplant 2012; 31: 611–617.
- 10.
Takayama H, Soni L, Kalesan B, Truby LK, Ota T, Cedola S, et al. Bridge-to-decision therapy with a continuous-flow external ventricular assist device in refractory cardiogenic shock of various causes. Circ Heart Fail 2014; 7: 799–806.
- 11.
Tsukiya T, Mizuno T, Takewa Y, Tatsumi E, Taenaka Y. Preclinical study of a novel hydrodynamically levitated centrifugal pump for long-term cardiopulmonary support: In vivo performance during percutaneous cardiopulmonary support. J Artif Organs 2015; 18: 300–306.
- 12.
Kishimoto S, Takewa Y, Tsukiya T, Mizuno T, Date K, Sumikura H, et al. Novel temporary left ventricular assist system with hydrodynamically levitated bearing pump for bridge to decision: Initial preclinical assessment in a goat model. J Artif Organs 2018; 21: 23–30.
- 13.
Fukushima N, Tatsumi E, Seguchi O, Takewa Y, Hamasaki T, Onda K, et al. Assessment of safety and effectiveness of the extracorporeal continuous-flow ventricular assist device (BR16010) use as a bridge-to-decision therapy for severe heart failure or refractory cardiogenic shock: Study protocol for single-arm non-randomized, uncontrolled, and investigator-initiated clinical trial. Cardiovasc Drugs Ther 2018; 32: 373–379.
- 14.
Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989; 10: 1–10.
- 15.
Kirklin JK, Naftel DC, Pagani FD, Kormos RL, Stevenson LW, Blume ED, et al. Seventh INTERMACS annual report: 15,000 patients and counting. J Heart Lung Transplant 2015; 34: 1495–1504.

