Abstract
Background:
To confirm the safety and efficacy of the IN.PACT Admiral drug-coated balloon (DCB) based on the indication approved by the Pharmaceuticals and Medical Devices Agency Japan in real-world patients with femoropopliteal artery disease.
Methods and Results:
IN.PACT PMS Japan was a prospective, multicenter, single-arm, post-market surveillance (PMS) study conducted in Japan that enrolled 304 participants (mean age 75.3±7.9 years). The primary endpoint was primary patency at 6 months following the index procedure, defined as freedom from clinically driven target lesion revascularization (CD-TLR) and freedom from restenosis as determined by duplex ultrasound (DUS) peak systolic velocity ratio (PSVR) ≤2.4 (assessed by the independent DUS core laboratory). Secondary endpoints included acute outcomes, primary patency at 12 months post-index procedure, freedom from CD-TLR, and major adverse events at 12 months. The mean lesion length was 97.81±58.97 mm. The primary endpoint, 6-month primary patency, was 91.3% (240/263). Kaplan-Meier estimates of primary patency and freedom from CD-TLR through 12 months were 91.5% and 94.1%, respectively. The CD-TLR rate was 5.8% (14/240) with low rates of thrombosis (0.8%) and target limb amputation (0.4%) at 12 months.
Conclusions:
The results of this real-world PMS study were consistent with outcomes from previous IN.PACT DCB studies, confirming the safety and efficacy of the IN.PACT Admiral DCB for broader use in patients seen in everyday practice.
The paclitaxel-based drug-coated balloon (DCB) has become an attractive revascularization option because of its anti-restenotic potential and “leave nothing or less behind” strategy that circumvents the use of metallic scaffolds, preserving future therapeutic options. Multiple randomized controlled trials (RCTs) and prospective nonrandomized studies have demonstrated the safety and effectiveness of DCBs for the treatment of individuals with symptomatic peripheral artery disease (PAD).1–12
However, most of the studies were conducted in Europe or the USA and studies on the real-world use of DCBs in ethnic populations are limited.
Editorial p ????
Three prospective studies – the IN.PACT SFA RCT, a large randomized trial conducted as phase I and II in Europe and the USA,1,3–5
the MDT-2113 SFA (also referred as IN.PACT SFA Japan) RCT conducted in Japan,13,14
and the single-arm IN.PACT SFA China15
– have demonstrated the safety and effectiveness of the IN.PACT Admiral DCB (Medtronic) for treating femoropopliteal occlusive disease in individuals with symptoms of claudication and/or ischemic rest pain (Rutherford Category [RC]).2–4
A complementary single-arm global study further expanded the performance of this DCB in a real-world population with longer, more complex lesions.11,16,17
The current post-market surveillance (PMS) study (IN.PACT PMS Japan) aimed to confirm the safety and effectiveness of this DCB based on the indication approved by the Pharmaceuticals and Medical Devices Agency (PMDA) Japan in a real-world population.
Methods
Study Design
IN.PACT PMS Japan was a prospective, multicenter, non-randomized, single-arm, surveillance study designed to confirm the safety and efficacy of the IN.PACT Admiral DCB based on the indication approved by Japan PMDA in real-world individuals with femoropopliteal artery disease. This DCB is indicated for percutaneous transluminal angioplasty of de novo and nonstented restenotic lesions with length ≤200 mm in superficial femoral and popliteal arteries with reference vessel diameters of ≥4 mm and ≤7 mm. Although there were no exclusion criteria set for this study, the guideline for proper use of the product based on the indication approved by Japan PMDA was followed (Supplementary Table 1).
Clinical follow-up was conducted at 1, 6 and 12 months post-index procedure. If a patient required target lesion revascularization (TLR) on a lesion that was treated using the IN.PACT Admiral DCB at the index procedure, those patients were followed up to 24 months (additional follow-up) post-index procedure. Clinical symptoms, major adverse events (MAEs), and vessel patency by duplex ultrasound (DUS) were assessed at each visit. Product compliance was reviewed by an Executive Committee comprised of 5 specialist physicians (including cardiovascular, surgical and radiology specialists). An independent DUS (VasCore; Massachusetts General Hospital, Boston, MA, USA) core laboratory analyzed the procedural and follow-up images. This PMS study was required and regulated by the PMDA and conducted in accordance with the Declaration of Helsinki, good clinical practice guidelines and Japanese Good Post-market Surveillance Practice (GPSP) regulations. Based on the ordinance of GPSP, patient agreement on participation in the surveillance is not required; however, participant consent is necessary to transmit personal data to overseas core laboratories, and for publications.
Medical Therapy
Medical therapy was provided as per institutional standards of care and the device’s Instructions for Use,18
which recommended that dual antiplatelet therapy be administered before the procedure and for a minimum of 1 month after the intervention, and that aspirin be continued for a minimum of 6 months after the procedure. Prolonged antiplatelet therapy could be given at the discretion of the physician.
Endpoints and Definitions
The primary endpoint was primary patency at 6 months following the index procedure, defined as freedom from clinically driven TLR (CD-TLR) and freedom from restenosis as determined by DUS peak systolic velocity ratio (PSVR) ≤2.4 (assessed by the independent DUS core laboratory). CD-TLR was defined as any re-intervention at the target lesion due to symptoms or decrease in ABI or TBI of ≥20% or >0.15 when compared with the post-procedure baseline ankle–brachial index (ABI) or TBI. Secondary endpoints included primary patency at 12 months post-index procedure, freedom from CD-TLR, MAE composite and its individual components [all-cause death, clinically driven target vessel revascularization (CD-TVR), major target limb amputation, and thrombosis at the target lesion site] at 12 months.
Additional assessments include device success, procedure success, clinical success, and improvement in RC. Device success was defined as successful delivery, inflation, deflation and retrieval of the intact study balloon device without bursting below the rated burst pressure. Procedural success was defined as residual stenosis ≤50% (nonstented subjects) or ≤30% (stented subjects) by physician’s visual assessment. Clinical success was defined as procedural success without procedural complications (death, major target limb amputation, thrombosis of the target lesion, or TVR) prior to discharge.
Patient Population
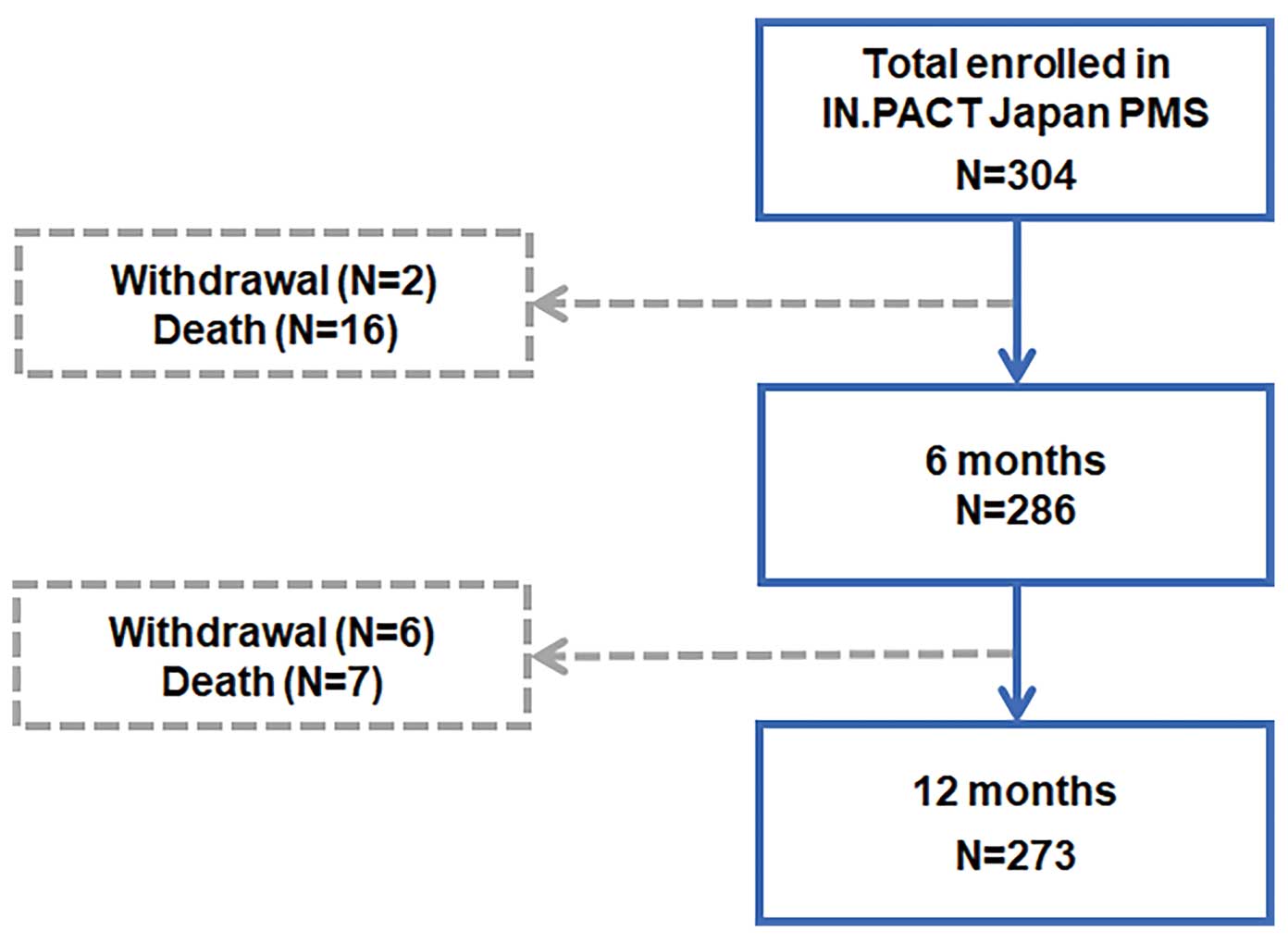
The 304 participants (mean age 75.3±7.9 years; 64.5% men) were enrolled at 19 centers in Japan. Initially, the study was set for 20 sites, but the required enrollment numbers were achieved before the last site was set up. Participant flow through the 12-month follow-up is described in
Figure 1.
Statistical Analysis
All analyses were based on the intention-to-treat principle. All summaries were based on non-missing assessments. Unless otherwise specified, all baseline demographics and clinical characteristics were summarized on a subject basis; lesion characteristics were summarized on a lesion basis; device success was analyzed on a device basis. For baseline characteristics, continuous variables are described as mean±standard deviation; dichotomous and categorical variables are described as counts and proportions. For assessment of the change in RC at 6 or 12 months, subjects were required to have data at baseline and 6 or 12 months. For MAE rates that were expressed as a proportion at 6 months or 12 months, the number of subjects with the corresponding event within 180 or 360 days was the numerator, and the numerator plus the number of subjects who had been in the study for at least 150 or 330 days with no event was the denominator. In addition, the Kaplan-Meier method was used to evaluate time-to-event data for primary patency, freedom from CD-TLR and freedom from death over the 12-month follow-up period. Estimates are presented with the 95% confidence interval (CI) where applicable. The primary patency through 12 months in subgroups RC 1–3 and RC 4–6 was also analyzed using the Kaplan-Meier method. The difference in the survival curves across subgroups was assessed using the log-rank test. The statistical significance level between subgroups was set at P<0.05. Statistical analyses were performed using SAS (version 9.4; SAS Institute, Cary, NC, USA).
Results
Baseline demographics and lesion characteristics of the IN.PACT PMS Japan full cohort are reported in
Table 1. In this cohort, participants were older (mean age >75 years) and presented with numerous comorbidities, including diabetes mellitus (62.2%), hypertension (80.3%), chronic renal insufficiency (44.7%), and coronary artery disease (54.6%). The mean lesion length was 97.81±58.97 mm, percent diameter stenosis was 83.77±14.90%, and 13.2% of lesions had total occlusion (100% stenosis). Calcified lesions were also substantial (62.6%), including 8.0% severely calcified. Provisional stenting was required for only 1 lesion due to a flow-limiting dissection [0.3% (1/364)]. The mean nominal paclitaxel dose received was 9,760.1±4,793.9 µg.
Table 1.
Participant Baseline and Lesion Characteristics
| Item |
IN.PACT PMS Japan (n=304 participants)
(n=364 lesions) (n=480 balloons) |
| Patient characteristics |
| Age (years) |
75.3±7.9 (304) |
| Sex |
| Female |
35.5% (108/304) |
| Male |
64.5% (196/304) |
| Smoking history |
60.2% (183/304) |
| Hyperlipidemia |
52.6% (160/304) |
| Diabetes |
62.2% (189/304) |
| Hypertension |
80.3% (244/304) |
| Chronic renal insufficiency |
44.7% (136/304) |
| Remain on dialysis |
34.5% (105/304) |
| Coronary artery diseasea |
54.6% (166/304) |
| ABI |
0.705±0.188 (280) |
| Lesion characteristics |
| Target lesion location |
| SFA |
84.6% (308/364) |
| Popliteal artery |
21.2% (77/364) |
| Diameter stenosis (%) |
83.77±14.90 (364) |
| Total lesion length (mm) |
97.81±58.97 (362) |
| Occluded lesion length (mm) |
12.29±35.12 (327) |
| RVD (mm) |
5.29±3.22 (359) |
| Calcification |
| None |
37.4% (130/348) |
| Mild to moderate |
39.4% (137/348) |
| Moderate to severe |
15.2% (53/348) |
| Severe calcification |
8.0% (28/348) |
| Total occlusion (100% stenosis) |
13.2% (48/364) |
| Procedural characteristics |
| Dissection present |
| No |
23.8% (114/479) |
| A |
18.4% (88/479) |
| B |
47.8% (229/479) |
| C |
9.2% (44/479) |
| D |
0.8% (4/479) |
| E |
0.0% (0/479) |
| F |
0.0% (0/479) |
| Provisional stent use per target lesion |
0.3% (1/364) |
| Provisional stent use per subject |
0.3% (1/304) |
| Nominal paclitaxel dose (μg)b |
9,760.1±4,793.9 |
Continuous data are presented as the mean±standard deviation; categorical data are presented as percentage (count/sample). Site reported data. aCoronary artery disease includes congestive heart failure and/or ischemic heart disease. bNominal paclitaxel dose was calculated as described previously.23 ABI, ankle-brachial index; RVD, reference vessel diameter; SFA, superficial femoral artery.
Acute outcomes of the full cohort are reported in
Table 2. Procedural success, defined as residual stenosis ≤50% (nonstented subjects) or ≤30% (stented subjects) by physician’s visual assessment, was achieved in 100% of subjects in the DCB group. There were no device-related adverse events and 1 potential procedure-related adverse events through to discharge.
Table 2.
Acute Outcomes
| |
IN.PACT PMS Japan (n=304 participants)
(n=364 lesions) (n=480 balloons) |
| Device successa |
98.1% (471/480) |
| Procedural successb |
100.0% (364/364) |
| Clinical successc |
98.0% (298/304) |
Categorical data are presented as percentage (count/sample). aDevice success defined as successful delivery, inflation, deflation and retrieval of the intact study balloon device without burst below the rated burst pressure (RBP), per DCB. bProcedural success defined as residual stenosis of ≤50% (nonstented subjects) or ≤30% (stented subjects) by physician’s visual assessment, per lesion. cClinical success defined as procedural success without procedural complications (death, major target limb amputation, thrombosis of the target lesion, or target vessel revascularization) prior to discharge, per subject.
The primary patency (freedom from CD-TLR and freedom from restenosis) rates at 6 and 12 months were 91.3% (240/263) and 82.6% (138/167), respectively (Table 3). The Kaplan-Meier estimate of primary patency was 91.5% (95% CI: 87.1% to 95.8%) and freedom from CD-TLR was 94.1% (95% CI: 90.4% to 97.8%) through 12 months (Figure 2A,B). The sharp tailing off in the primary patency curve was expected due to more events being reported around scheduled visits when DUS was assessed (±30 days), and near the study end period, the curve tends to drop further due to the censored data mainly of patients who had completed the study and exited.
Table 3.
Primary and Secondary Outcomes
| |
IN.PACT PMS Japan
(n=304 participants) |
| Primary patency at 6-month post-index procedurea |
91.3% (240/263) |
| 95% confidence interval |
[87.2%, 94.4%] |
| Freedom from CD-TLRb |
96.9% (254/262) |
| Freedom from restenosis determined by core laboratory assessment |
94.7% (266/281) |
| Primary patency at 12-month post-index procedurea |
82.6% (138/167) |
| 95% confidence interval |
[76.0%, 88.1%] |
| Freedom from CD-TLRb |
91.4% (148/162) |
| Freedom from re-stenosis determined by core laboratory assessment |
93.4% (211/226) |
| Cumulative complications within 6 months |
| MAE composite |
9.0% (26/289) |
| All-cause death |
5.5% (16/289) |
| Target limb amputation |
0.3% (1/289) |
| CD-TVR revascularization |
2.8% (8/289) |
| Thrombosis |
0.7% (2/289) |
| CD-TLR |
2.1% (6/289) |
| Any TLR |
2.4% (7/289) |
| Cumulative complications within 12 months |
| MAE composite |
17.1% (41/240) |
| All-cause death |
9.6% (23/240) |
| Target limb amputation |
0.4% (1/240) |
| CD-TVR revascularization |
7.1% (17/240) |
| Thrombosis |
0.8% (2/240) |
| CD-TLR |
5.8% (14/240) |
| Any TLR |
6.7% (16/240) |
Categorical data are presented as percentage (count/sample). aDefined as freedom from CD-TLR and freedom from restenosis as determined by DUS peak systolic velocity ratio ≤2.4 (assessed by the independent DUS core laboratory). bDefined as any re-intervention at the target lesion due to symptoms or decrease in the ABI or TBI ≥20% or >0.15 when compared with post-procedure baseline ABI or TBI. ABI, ankle-brachial index; CD-TLR, clinically driven target lesion revascularization; CD-TVR, clinically driven target vessel revascularization; DUS, duplex ultrasound; MAE, major adverse event; TLR, target lesion revascularization; TVR, target vessel revascularization.

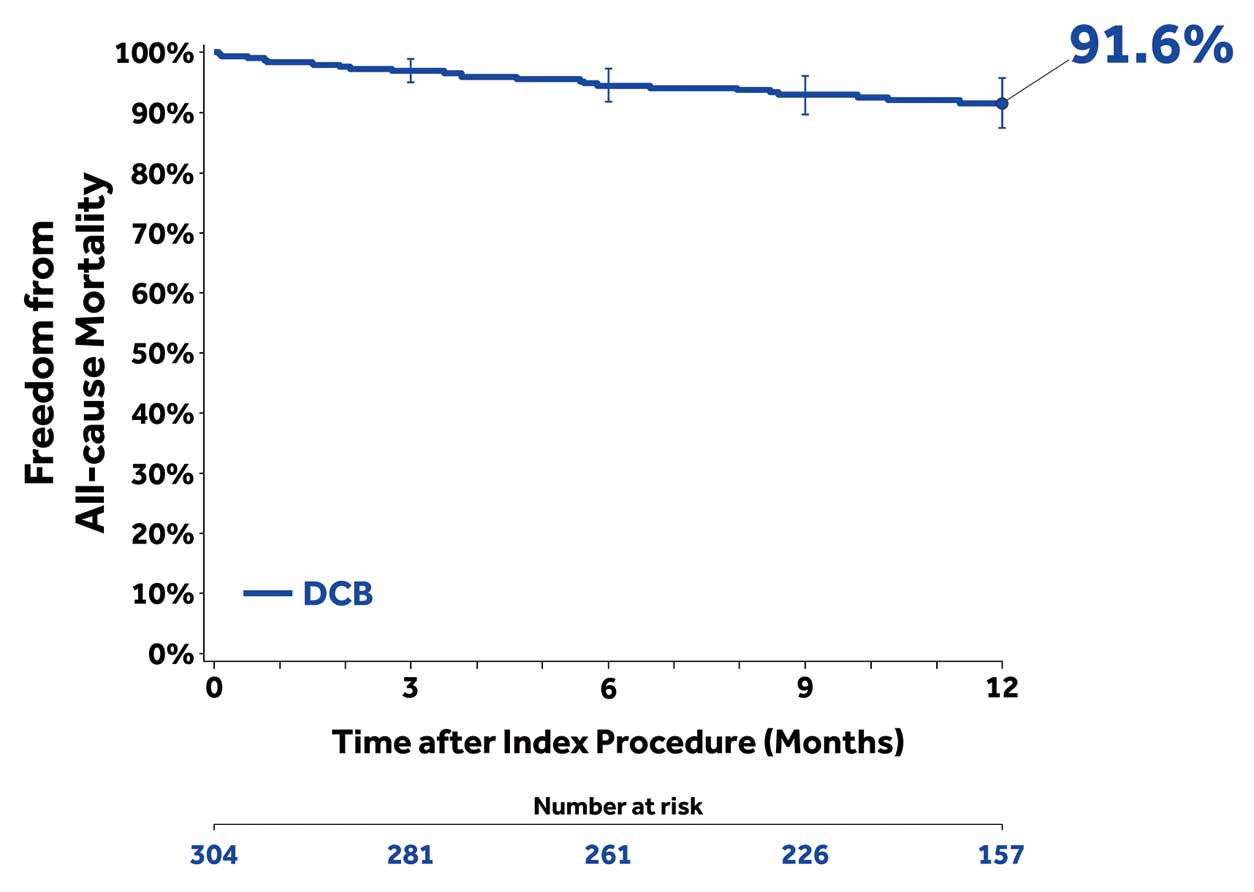
The MAEs composite [all-cause death, CD-TVR, major target limb amputation, and thrombosis at the target lesion site] was 17.1% at 12 months (Table 3). There were 23 deaths (9.6%) and 1 target limb amputation (0.4%) within the 12-month follow-up. The Kaplan-Meier estimate of freedom from all-cause death through 12 months was 91.6% (Figure 3). The rate of thrombosis was 0.8% (2/240) and 1 target limb amputation (0.4%) occurred within the 12-month follow-up. The rate of CD-TLR was 5.8% (14/240) at 12 months.
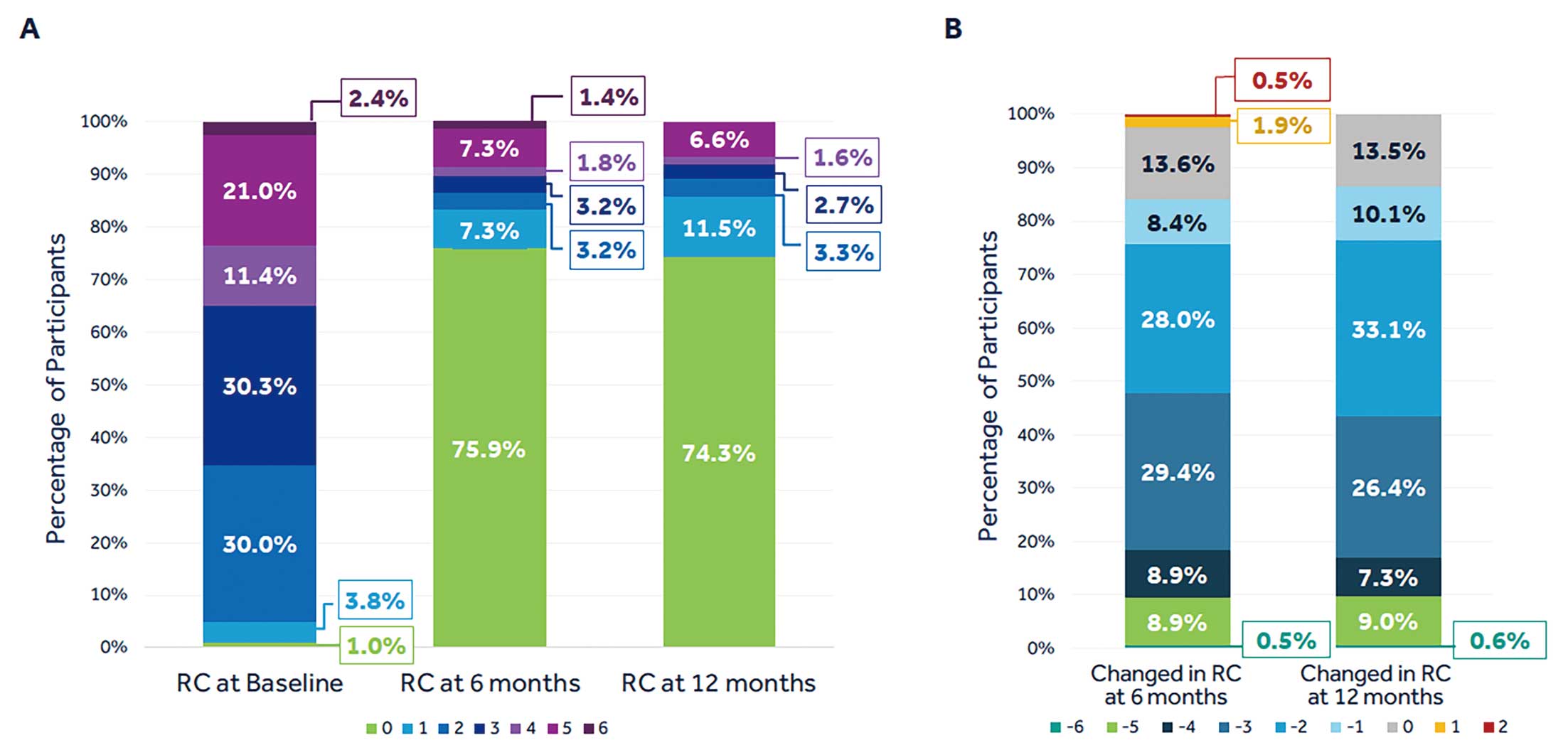
At both 6 and 12 months, participants showed improvement in RC compared with baseline (Figure 4). The majority of participants were asymptomatic (74.3%), and 86.5% of participants showed improvement of ≥1 RC at 12 months.
Subgroup Analyses
Baseline demographic and lesion characteristics of participants stratified by their RC (RC 1–3 vs. RC 4–6) are reported in
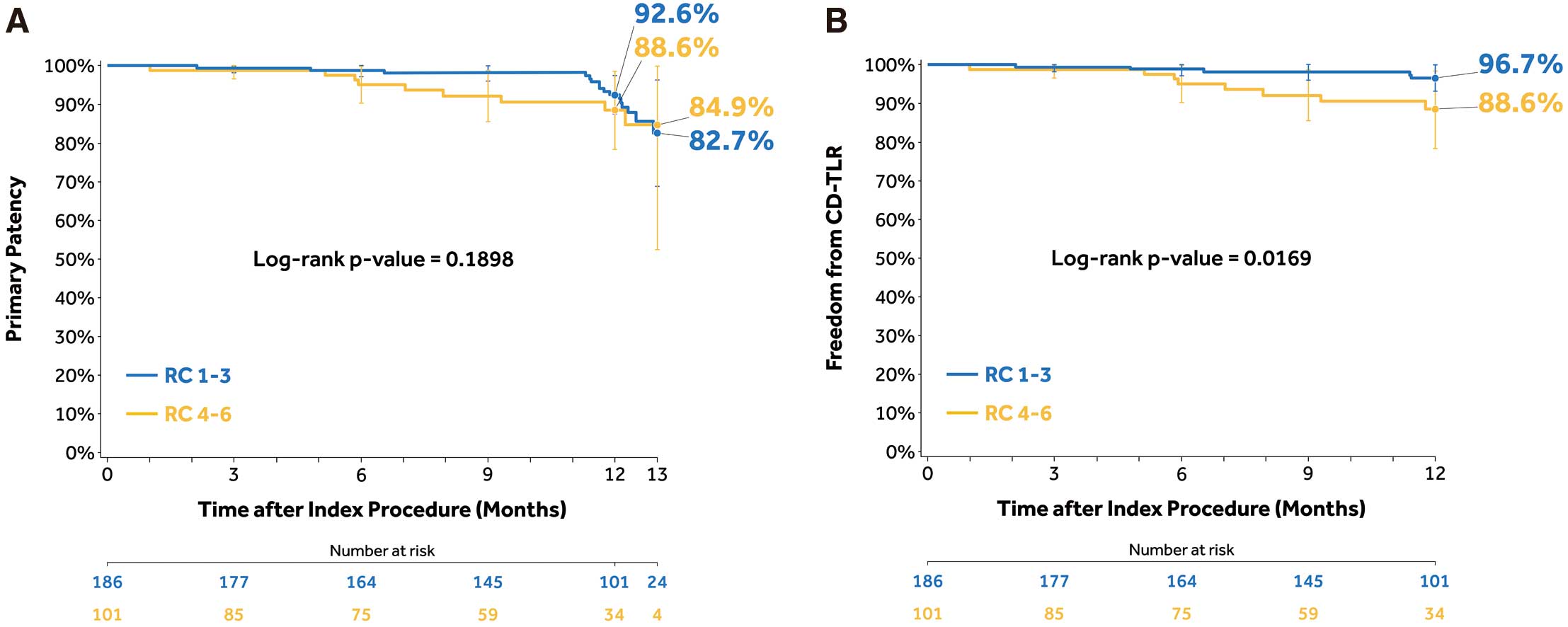
Supplementary Table 2. Participants in the RC 4–6 group included more women (49.5% vs. 29.6%), had higher rates of diabetes (69.3% vs. 59.7%) and chronic renal insufficiency (61.4% vs. 36.0%) when compared with the RC 1–3 group. The Kaplan-Meier estimate of primary patency through 12 months was 88.6% vs. 92.6% (log-rank P=0.1898) in the RC 4–6 vs. RC 1–3 group (Figure 5A). Through 12 months, freedom from CD-TLR was lower in the RC 4–6 compared with the RC 1–3 group (88.6% vs. 96.7%; log-rank P=0.0169 (Figure 5B). The all-cause death rates at 12 months were 6.0% (9/151) and 17.3% (13/75) in the RC 1–3 and RC 4–6 groups, respectively. There was only 1 target limb amputation in the RC 4–6 group (1.3% [1/75]) and none in the RC 1–3 group at 12 months.
Discussion
The IN.PACT SFA Japan, an investigational device exemption (IDE) randomized trial, has established the superiority of the paclitaxel-coated IN.PACT Admiral DCB compared with PTA for femoropopliteal occlusive disease through 3 years in a Japanese population.13,14,19
Results of the present PMS study (IN.PACT PMS Japan) further confirmed the safety and efficacy of the IN.PACT Admiral DCB in a real-world patient population in Japan and supported the previous findings.
The present study had no exclusion criteria and thus represented the broader patient population seen in everyday practice, with a high incidence of traditional risk factors including older age (mean age >75 years), diabetes mellitus, hypertension, coronary artery disease, chronic renal insufficiency, and chronic limb-threatening ischemia (CLTI). Despite the complexity of the patients’ characteristics, the 12-month primary patency of 91.5% in the IN.PACT PMS Japan (mean lesion length 9.8 cm) was favorable compared with the 12-month primary patency of DCB studies conducted in Europe and the USA: the IN.PACT SFA (87.5%; mean lesion length 8.9 cm);5
LEVANT 2 (73.5%; mean lesion length 6.3 cm);2
ILLUMENATE European (89.0%; mean lesion length 7.2 cm);20
and ILLUMENATE Pivotal (82.3%; mean lesion length 8.0 cm).7
This finding is noteworthy, considering that the mean lesion length was much longer, and patients’ clinical characteristics were more complex in the IN.PACT PMS Japan study compared with the other DCB studies. Other real-world DCB studies have also reported DCB outcomes for femoropopliteal lesions, although participant populations were predominantly Caucasian in those studies. Nonetheless, the 12-month freedom from CD-TLR of 94.1% in the IN.PACT PMS Japan study was comparable to these studies: 12-month freedom from CD-TLR of 92.6% in the IN.PACT Global Study (mean lesion length 12.1 cm);11
94.1% in the Lutonix Global SFA registry (mean lesion length 10.1);10
94.8% in the ILLUMENATE Global study (mean lesion length 7.5);12
and 93.9% in the femoropopliteal cohort of the BIOLUX P-III Global registry (mean lesion length 9.0 cm).21
Although the mean lesion length in the IN.PACT PMS Japan (9.8 cm) was within the range of these other real-world studies (7.5–12.1 cm), participants in the present study were older and presented with more CLTI, except for the BIOLUX P-III Global registry.
There are limited DCB studies that report outcomes of exclusively Asian populations. The performance outcomes of the DCB in the present study (primary patency of 91.5% and freedom from CD-TLR of 94.1%) were consistent with the DCB arm of the IN.PACT SFA Japan outcomes (primary patency of 93.9% and freedom from CD-TLR of 97.1%).13
The IN.PACT PMS Japan had a higher incidence of chronic renal insufficiency (44.7% vs. 9.0%), more advanced PAD, slightly longer lesion length (9.8 cm vs. 9.2 cm), as well as more popliteal involvement (15.2% vs. 1%) compared with IN.PACT SFA Japan. The results of the IN.PACT PMS Japan were also favorable when compared with primary patency of 86.4% and freedom from CD-TLR of 91.0% in the Zilver PTX PMS study in Japan; furthermore, not leaving a permanent metallic scaffold within the vessel bed may be advantageous for future procedures.
Another interesting finding of the present study was that angioplasty with the DCB was consistently effective even in patients with CLTI, as shown by the findings that the 12-month primary patency rates were not significantly different between the RC 4–6 and RC 1–3 groups. Freedom from CD-TLR was also high in the RC 4–6 group, although it was lower than the RC 1–3 group. These findings are vital considering the very limited number of studies that specifically reports outcomes of DCB angioplasty in real-world CLTI patients.
Recently, the safety of paclitaxel-coated devices has come into question.22
In the present study, there were no safety concerns; no device-related adverse events were reported. The mean paclitaxel dose (9,760.1±4,793.9 µg) in the present analysis is similar to that reported previously for femoropopliteal DCB interventions.23
In the present study, only 1 patient required a major amputation and 2 cases of thrombosis occurred through 6 months, and no further cases of amputation or thrombosis through 12 months. These findings are remarkable, given the high amputation rates in CLTI patients [1-year rate of 15–20%24]. The 1-year all-cause mortality rate of 9.6% at 12 months in the present study was higher than reported for IDE trials, IN.PACT SFA Japan (0.0%) and IN.PACT SFA China (2.9%), which was expected because the patient population in the IN.PACT PMS Japan was older, sicker and included substantial number of CLTI cases. A multicenter retrospective analysis of CLTI patients (n=995) in Japan reported that the mortality rate in this patient population was as high as 41% in 2 years, and age was an independent risk factor for death.25
Another study reported a 12-month survival rate of 86.1% (13.9% cumulative incidence of death) in CLTI patients who had undergone endovascular therapy.26
Mueller et al also reported a substantial variation in mortality rate among patients with PAD by their age and diabetic status: lowest for nondiabetic patients aged <75 years (5-year mortality rate of 10%) to highest for diabetic patients aged ≥75 years (5-year mortality rate of 52%).27
Study Limitations
Relevant limitations of this study include the single-arm study design without a comparator and no clinical events committee to adjudicate revascularization and MAEs. Although the 6-month follow-up visit compliance was good, the 12-month follow-up visit compliance was lower than that of the IDE study (IN.PACT SFA Japan). The strengths of the study include core laboratory adjudication of procedural and follow-up images, a broad patient population that represents a real-world scenario, and a subgroup analysis of data from CLTI cases, a challenging patient population.
Conclusions
Results from this real-world PMS study were consistent with the IN.PACT SFA Japan and other IN.PACT studies, confirming the safety and efficacy of the IN.PACT Admiral DCB for broader use in patients seen in everyday practice.
Acknowledgments
The authors thank the participants for their involvement in this study. The authors also recognize the principal investigators and institutions in Japan that enrolled participants in the study: Kenji Ando, MD, Kokura Memorial Hospital, Tokyo, Japan; Toshiaki Mano, MD, Kansai Rosai Hospital, Hyogo, Japan; Yoshisato Shibata, MD, Miyazaki Medical Association Hospital, Miyazaki, Japan; Kan Zen, MD, University Hospital, Kyoto Prefectural University of Medicine, Kyoto, Japan; Shigeo Ichihashi, MD, Nara Medical University Hospital, NR, Japan; Shigeru Nakamura, MD, Kyoto Katsura Hospital, Tokyo, Japan; Yoshiaki Yokoi, MD, Kishiwada Tokushukai Hospital, Osaka, Japan; Hiroshi Ando, MD, Kasukabe Chuo General Hospital, Saitama, Japan; Naoto Inoue, MD, Sendai Kousei Hospital, MG, Japan; Keisuke Hirano, MD, and Shinsuke Mori, MD, Saiseikai Yokohamashi Tobu Hospital, Kanagawa, Japan; Takuo Nakagami, MD, Omihachiman Community Medical Center, Shiga, Japan; and Yoshito Yamamoto, MD, Iwaki City Medical Center, Fukushima, Japan. The authors also thank Kathleen Cahill, MS and Hiroko Ookubo, degree, for technical review; Sangeeta Yendrembam, PhD, for medical writing assistance in accordance with Good Publication Practice guidelines (http://www.ismpp.org/gpp3).
Disclosures
Y.S., O.I., M.F., D.K., S.S., K.U. and H.Y. have no conflicts of interest; M.N. received honoraria from Bard, Boston Scientific and Medtronic; J.G. and E.J.F. are full-time salaried employees of Medtronic.
Funding
Medtronic sponsored the trial and owns the data. The authors had full access to all the data, interpretation, manuscript writing and had full and final responsibility for the decision to submit for publication. Authors received no specific funding for this work or preparation of the manuscript.
IRB Iinformation
The present study was approved by the Institutional Review Board, Kasukabe Chuo General Hospital (reference no. 1801), and institutional review board or ethics committee at the sites reviewed and approved the study protocol or exempted as applicable.
Data Availability
The results, deidentified participant data, and related documents will be shared on a request basis and upon approval from the sponsor. The data will become available 1 year after completion of the study. The study sponsor (Medtronic) evaluates on a study-by-study basis whether there is an opportunity to share clinical trial data with qualified scientific or medical researchers, consistent with the associated informed consent and applicable laws and regulations. Data will be provided after approval according to sponsor policies.
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-21-0491
References
- 1.
Tepe G, Laird J, Schneider P, Brodmann M, Krishnan P, Micari A, et al. Drug-coated balloon versus standard percutaneous transluminal angioplasty for the treatment of superficial femoral and popliteal peripheral artery disease: 12-month results from the IN.PACT SFA randomized trial. Circulation 2015; 131: 495–502.
- 2.
Rosenfield K, Jaff MR, White CJ, Rocha-Singh K, Mena-Hurtado C, Metzger DC, et al. Trial of a paclitaxel-coated balloon for femoropopliteal artery disease. N Engl J Med 2015; 373: 145–153.
- 3.
Laird JR, Schneider PA, Tepe G, Brodmann M, Zeller T, Metzger C, et al. Durability of treatment effect using a drug-coated balloon for femoropopliteal lesions: 24-month results of IN.PACT SFA. J Am Coll Cardiol 2015; 66: 2329–2338.
- 4.
Schneider PA, Laird JR, Tepe G, Brodmann M, Zeller T, Scheinert D, et al. Treatment effect of drug-coated balloons is durable to 3 years in the femoropopliteal arteries: Long-term results of the IN.PACT SFA randomized trial. Circ Cardiovasc Interv 2018; 11: e005891.
- 5.
Laird JA, Schneider PA, Jaff MR, Brodmann M, Zeller T, Metzger DC, et al. Long-term clinical effectiveness of a drug-coated balloon for the treatment of femoropopliteal lesions. Circ Cardiovasc Interv 2019; 12: e007702.
- 6.
Tepe G, Schnorr B, Albrecht T, Brechtel K, Claussen CD, Scheller B, et al. Angioplasty of femoral-popliteal arteries with drug-coated balloons: 5-year follow-up of the THUNDER trial. JACC Cardiovasc Interv 2015; 8: 102–108.
- 7.
Krishnan P, Faries P, Niazi K, Jain A, Sachar R, Bachinsky WB, et al. Stellarex Drug-coated balloon for treatment of femoropopliteal disease: Twelve-month outcomes from the randomized ILLUMENATE pivotal and pharmacokinetic studies. Circulation 2017; 136: 1102–1113.
- 8.
Scheinert D, Schulte KL, Zeller T, Lammer J, Tepe G. Paclitaxel-releasing balloon in femoropopliteal lesions using a BTHC excipient: Twelve-month results from the BIOLUX P-I randomized trial. J Endovasc Ther 2015; 22: 14–21.
- 9.
Jia X, Zhang J, Zhuang B, Fu W, Wu D, Wang F, et al. Acotec drug-coated balloon catheter: Randomized, multicenter, controlled clinical study in femoropopliteal arteries: Evidence from the AcoArt I Trial. JACC Cardiovasc Interv 2016; 9: 1941–1949.
- 10.
Thieme M, Von Bilderling P, Paetzel C, Karnabatidis D, Perez Delgado J, Lichtenberg M, et al. The 24-month results of the Lutonix global SFA registry: Worldwide experience with Lutonix drug-coated balloon. JACC Cardiovasc Interv 2017; 10: 1682–1690.
- 11.
Zeller T, Brodmann M, Micari A, Keirse K, Peeters P, Tepe G, et al. Drug-coated balloon treatment of femoropopliteal lesions for patients with intermittent claudication and ischemic rest pain. Circ Cardiovasc Interv 2019; 12: e007730.
- 12.
Schroe H, Holden AH, Goueffic Y, Jansen SJ, Peeters P, Keirse K, et al. Stellarex drug-coated balloon for treatment of femoropopliteal arterial disease-The ILLUMENATE Global Study: 12-Month results from a prospective, multicenter, single-arm study. Catheter Cardiovasc Interv 2018; 91: 497–504.
- 13.
Iida O, Soga Y, Urasawa K, Saito S, Jaff MR, Wang H, et al. Drug-Coated Balloon vs. Standard percutaneous transluminal angioplasty for the treatment of atherosclerotic lesions in the superficial femoral and proximal popliteal arteries: One-year results of the MDT-2113 SFA Japan randomized trial. J Endovasc Ther 2018; 25: 109–117.
- 14.
Iida O, Soga Y, Urasawa K, Saito S, Jaff MR, Wang H, et al. Drug-coated balloon versus uncoated percutaneous transluminal angioplasty for the treatment of atherosclerotic lesions in the superficial femoral and proximal popliteal artery: 2-year results of the MDT-2113 SFA Japan randomized trial. Catheter Cardiovasc Interv 2019; 93: 664–672.
- 15.
Chen Z, Guo W, Jiang W, Wang F, Fu W, Zou Y, et al. IN.PACT SFA Clinical study using the IN.PACT Admiral drug-coated balloon in a Chinese patient population. J Endovasc Ther 2019; 26: 471–478.
- 16.
Micari A, Brodmann M, Keirse K, Peeters P, Tepe G, Frost M, et al. Drug-coated balloon treatment of femoropopliteal lesions for patients with intermittent claudication and ischemic rest pain: 2-year results from the IN.PACT global study. JACC Cardiovasc Interv 2018; 11: 945–953.
- 17.
Torsello G, Stavroulakis K, Brodmann M, Micari A, Tepe G, Veroux P, et al. Three-year sustained clinical efficacy of drug-coated balloon angioplasty in a real-world femoropopliteal cohort. J Endovasc Ther 2020; 27: 693–705.
- 18.
IN.PACTTM AdmiralTM instructions for use. https://www.info.pmda.go.jp/downfiles/md/PDF/530366/530366_22900BZX00295000_A_01_01.pdf (accessed November 12, 2020).
- 19.
Soga Y, Iida O, Urasawa K, Saito S, Jaff MR, Wang H, et al. Three-year results of the IN.PACT SFA Japan trial comparing drug-coated balloons with percutaneous transluminal angioplasty. J Endovasc Ther 2020; 27: 946–955.
- 20.
Schroeder H, Werner M, Meyer DR, Reimer P, Kruger K, Jaff MR, et al. Low-dose paclitaxel-coated versus uncoated percutaneous transluminal balloon angioplasty for femoropopliteal peripheral artery disease: One-year results of the ILLUMENATE European randomized clinical trial (Randomized Trial of a Novel Paclitaxel-Coated Percutaneous Angioplasty Balloon). Circulation 2017; 135: 2227–2236.
- 21.
Tepe G, Zeller T, Moscovic M, Corpataux JM, Christensen JK, Keirse K, et al. Paclitaxel-coated balloon for the treatment of infrainguinal disease: 12-month outcomes in the all-comers cohort of BIOLUX P-III global registry. J Endovasc Ther 2020; 27: 304–315.
- 22.
Katsanos K, Spiliopoulos S, Kitrou P, Krokidis M, Karnabatidis D. Risk of death following application of paclitaxel-coated balloons and stents in the femoropopliteal artery of the leg: A systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc 2018; 7: e011245.
- 23.
Schneider PA, Laird JR, Doros G, Gao Q, Ansel G, Brodmann M, et al. Mortality not correlated with paclitaxel exposure: An independent patient-level meta-analysis of a drug-coated balloon. J Am Coll Cardiol 2019; 73: 2550–2563.
- 24.
Duff S, Mafilios MS, Bhounsule P, Hasegawa JT. The burden of critical limb ischemia: A review of recent literature. Vasc Health Risk Manag 2019; 15: 187–208.
- 25.
Soga Y, Iida O, Takahara M, Hirano K, Suzuki K, Kawasaki D, et al. Two-year life expectancy in patients with critical limb ischemia. JACC Cardiovasc Interv 2014; 7: 1444–1449.
- 26.
Soga Y, Mii S, Aihara H, Okazaki J, Kuma S, Yamaoka T, et al. Comparison of clinical outcome after bypass surgery vs. endovascular therapy for infrainguinal artery disease in patients with critical limb ischemia. Circ J 2013; 77: 2102–2109.
- 27.
Mueller T, Hinterreiter F, Luft C, Poelz W, Haltmayer M, Dieplinger B. Mortality rates and mortality predictors in patients with symptomatic peripheral artery disease stratified according to age and diabetes. J Vasc Surg 2014; 59: 1291–1299.