Abstract
Background:
OPC-61815, a prodrug of tolvaptan, is an injectable aquaretic drug. This study evaluated the tolerability of OPC-61815 in patients with congestive heart failure (CHF) who had difficulty with, or were incapable of, oral intake in a multicenter, uncontrolled, open-label Phase III study.
Methods and Results:
Forty-five patients were enrolled at 30 Japanese sites. OPC-61815 infusion was administered once daily; the 8 mg initial dose could be increased to 16 mg if the dose escalation criteria were met. Patients were treated for up to 5 days. Thirty-eight patients maintained the 8-mg dose and 7 had a dose increase to 16 mg; 41 completed the trial (34 completed early). One patient had mild hypernatremia. No significant safety concerns were observed with OPC-61815 administration at a starting dose of 8 mg and with dose escalation in accordance with the protocol-specified criteria. Treatment resulted in weight decrease (−3.01 kg); improvement or disappearance rates for other CHF symptoms (including edema, dyspnea, orthopnea, pulmonary congestion, and rales) indicated that treatment was effective. Urine excretion was increased 0–1 h after OPC-61815 administration and reached a maximum level at 1–2 h.
Conclusions:
The tolerability of once daily (up to 5 days) intravenous OPC-61815 (8 mg or 16 mg) was confirmed in patients with CHF who had difficulty with, or were incapable of, oral intake.
The worldwide prevalence of heart failure (HF) in 2017 was estimated to be 64.34 million cases,1
and the associated disability is increasing as the global population continues to increase in number and age. This is particularly true for Japan, a country that has a rapidly growing number of individuals aged ≥65 years,2
and where the number of patients hospitalized for acute HF is rising each year.3
One of the main reasons for hospitalization of patients with HF is congestion.4,5
For these patients, loop diuretics are generally considered the cornerstone of treatment.6–9
Intravenous drugs are most commonly used, given the condition of patients at the time of hospitalization (i.e., administration of oral medication may not be possible or desirable) and the desire for a faster time to onset of drug effectiveness. However, patients may not respond to treatment or may develop resistance to diuretics over time.6–9
Loop diuretic use is also associated with potential side-effects including worsening renal function and electrolyte disturbances.9–11
Thus, additional treatment options are often necessary.
Editorial p ????
Tolvaptan is a vasopressin antagonist that promotes water excretion (aquaresis) without affecting electrolyte excretion.12
When administered as an oral aquaretic agent, tolvaptan has proven to be a useful clinical treatment option for HF patients with volume overload despite having received other conventional diuretics.13–15
In Japan, the number of patients receiving tolvaptan therapy has been increasing since its launch; in 2018, 39% of inpatients with HF were prescribed tolvaptan.16
However, tolvaptan is only available as oral formulations (tablet or granules) and is not readily water soluble.17–19
This places limitations on its use in several patient groups, including those unable to take oral medication or who have difficulty in drinking water due to impaired consciousness, ventilation, risk of aspiration, impaired swallowing reflex, or other factors. Based on the need for injectable aquaretic drugs for these patients, OPC-61815 was developed; this prodrug of tolvaptan has improved water solubility (Otsuka Pharmaceutical Co., Ltd.; data on file), and is readily metabolized to tolvaptan in the body following intravenous administration.
There is some concern that treatment with aquaretic agents such as OPC-61815 may increase the risk of hypernatremia and other diuretic-associated adverse events in patients who have difficulty in swallowing or drinking water compared with those who do not.18,20
Patients usually self-regulate their fluid balance by drinking water, but this may be difficult for patients with an impaired ability to swallow or those who experience a loss of consciousness associated with pulmonary edema. Moreover, subjective symptoms such as a dry mouth, which indicate thirst and influence the decision to drink fluids, may not be acted upon if patients are unaware of the symptoms or are unable to communicate them to caregivers. Fluid levels may be more difficult to manage in these patients, and appropriate fluid monitoring and management are necessary when patients with HF are being treated with OPC-61815.
Importantly, the recent Phase III non-inferiority study of OPC-61815 (ClinicalTrials.gov [NCT03962101]) used tolvaptan tablets as a comparator; as such, patients who had difficulty with fluid intake or those who were unable to sense thirst were excluded. Thus, it was considered necessary to conduct a separate clinical study to assess the tolerability of OPC-61815 in patients with congestive HF (CHF) who had difficulty with, or were incapable of, oral intake. Herein, we report the findings of a multicenter, uncontrolled, open-label Phase III trial that was conducted to confirm the tolerability of once-daily intravenous OPC-61815 (8 or 16 mg) for a maximum of 5 days in CHF patients who have volume overload despite having received injectable diuretics (other than vasopressin antagonists) and who had difficulty with, or were incapable of, oral intake (ToleRability of the Intravenously administered TOlvaptaN prodrug OPC-61815 in patients with congestive Heart Failure who have difficulty with, or are incapable of, oral intake [TRITON-HF]).
Methods
Patients
Patients were eligible to participate in the study if they were diagnosed with CHF with volume overload despite treatment with injectable diuretics; were currently receiving injectable loop diuretics (dose equivalent to furosemide 20 mg/day or higher); had lower limb edema, pulmonary congestion, or jugular venous distention due to volume overload; were aged 20–85 years at the time of consent; had difficulty with, or were incapable of, oral intake, including patients who needed fasting management, according to the judgment of the investigator/subinvestigator; and were currently hospitalized or were able to be hospitalized from the time of informed consent to the end of the treatment period. Major exclusion criteria were use of a ventricular assist device; medical conditions that could be adversely affected by aquaretic treatment (e.g., suspected hypovolemia, hypertrophic cardiomyopathy, valvular disease with significant valvular stenosis, or hepatic encephalopathy); medical history or diagnosis of acute myocardial infarction (within 30 days prior to screening), active myocarditis, amyloid cardiomyopathy, ventricular tachycardia or fibrillation (within 30 days prior to screening), cerebrovascular disease (within 6 months prior to screening), poorly controlled diabetes, anuria, dysuria associated with urinary tract obstruction or tumor, open heart surgery (within 30 days prior to screening), or hepatic impairment; history of hypersensitivity to any of the ingredients of OPC-61815 (including prior reactions to tolvaptan or benzazepines); supine systolic blood pressure <90 mmHg, serum/plasma creatinine >3 mg/dL, or serum/plasma sodium <125 mEq/L or >147 mEq/L; patients having difficulty with spontaneous respiration or who had tracheal intubation under sedation; and any other reason which, in the opinion of the investigator, could endanger the patients or confound study analyses. Only patients who were able to provide written informed consent prior to study participation were enrolled; patients with severe consciousness disturbances (i.e., coma or stupor) were ineligible.
Study Design and Treatment
This was a multicenter, uncontrolled, open-label trial to confirm the tolerability of intravenous OPC-61815 (8 or 16 mg once daily, 5 days maximum) in patients with CHF with volume overload despite treatment with injectable diuretics and who have difficulty with, or are incapable of, oral intake. This study was registered at ClinicalTrials.gov (NCT03962101) and ClinicalTrials.jp (JapicCTI-194715). The protocol, amendments, and the informed consent form were reviewed and approved by the governing institutional review board for each investigational trial site. This trial was conducted in accordance with the Declaration of Helsinki and was in compliance with the protocol, Good Clinical Practice guidelines, and all applicable local laws and regulatory requirements.
The study was conducted at 30 sites in Japan between June 17, 2019 and June 30, 2020, and consisted of a screening period, a treatment period (up to 6 days), and a follow-up period. Details of the study design can be found in
Supplementary Figure. OPC-61815 was administered once daily as a 1-h infusion (55–70 min allowed). Treatment was initiated with an 8-mg dose, and eligibility for dose escalation was assessed on Day 2 of the treatment period (and Day 3, if applicable). If the dose escalation criteria were met, the dose was increased to 16 mg; otherwise, the dose remained at 8 mg. Dose escalation criteria were as follows: inadequate response, serum sodium concentration of ≤147 mEq/L at 8 mg, no evident increase of >10 mEq/L in serum sodium concentration within 24 h of the start of OPC-61815 administration, and no safety concerns. An inadequate response on Day 2 was defined as: (1) the presence of any congestive symptoms (i.e., lower limb edema, hepatomegaly, jugular venous distension, cardiothoracic ratio, pulmonary rales, or cardiac third sound) and a daily urine volume increase of ≤500 mL (from Day 1 to Day 2); or (2) an increase of >500 mL that could either not be confirmed or was accompanied by no reductions in bodyweight and no improvements in congestive symptoms. On Day 3, an inadequate response was defined as a body weight that did not decrease when compared with Day 2 and no improvement in congestive symptoms. If a patient experienced treatment-emergent adverse events (TEAEs) associated with the aquaretic effect of OPC-61815 after the dose was increased to 16 mg, the dose was reduced to 8 mg and could not be re-escalated to 16 mg.
Patients continued receiving injectable loop diuretics as needed during the screening and treatment periods and were hospitalized from the time of informed consent to the day after the final administration of OPC-61815. Patients completed the trial early (prior to Day 5) if they became capable of fluid management by oral diuretics alone or if they had all congestive findings resolved, requiring no further improvement of volume overload.
Study Outcomes
Safety evaluations included TEAEs, clinical laboratory tests, physical examination, vital signs (blood pressure, pulse rate, and body temperature), and 12-lead electrocardiogram. Efficacy outcomes included change in body weight, congestive symptoms (lower limb edema, jugular venous distention, pulmonary rales, and pulmonary congestion), dyspnea (paroxysmal nocturnal dyspnea, orthopnea, and subject-assessed dyspnea), and New York Heart Association (NYHA) classification. Pharmacodynamic outcomes included daily urine volume and changes in serum sodium concentration, serum potassium concentration, serum osmolality, and biomarker levels (plasma arginine vasopressin [AVP], plasma renin activity, plasma B-type natriuretic peptide [BNP], serum N-terminal proBNP, and serum troponin I).
Statistical Methods
It was determined that at least 40 patients should be enrolled and receive the study drug to achieve the study objectives. For a target sample size of 40 patients, the probability of TEAEs occurring at an incidence of 5% and 4% was calculated to be 87% and 80%, respectively.
The safety analysis set included all patients who received at least 1 dose of OPC-61815, the efficacy analysis set consisted of all patients who received at least 1 dose of OPC-61815 and had post-treatment efficacy data, and the pharmacodynamic analysis set consisted of all patients who received at least 1 dose of OPC-61815 and had post-treatment pharmacodynamic data. Continuous and categorical data were summarized using descriptive statistics. For efficacy parameters, descriptive statistics were calculated for measured values and changes from baseline at the time of final OPC-61815 administration (body weight), improvement rate (lower limb edema, patient assessed dyspnea, pulmonary congestion, NYHA classification), and/or disappearance rate (lower limb edema, paroxysomal nocturnal dyspnea, orthopnea, pulmonary congestion, pulmonary rales). Patients with NYHA Class II and higher were included in the analysis for NYHA classification. TEAEs were evaluated in the safety analysis set and were coded using system organ class (SOC) and in the preferred term according to the Medical Dictionary for Regulatory Activities version 23.0. Values for serum creatinine, blood urea nitrogen, and biomarkers at baseline and on the day after final OPC-61815 administration were compared using a paired t-test (post-hoc analysis). Biomarker values had non-normal distributions and were log-transformed prior to performing the paired t-test. Here, a P value of <0.05 was considered as statistically significant.
All analyses were conducted for the total population and according to whether the patient had dose escalation. All study calculations were performed using SAS software version 9.4 M5 (SAS Institute, Inc., Cary, NC, USA).
Results
Patients
Patient disposition is shown in
Figure 1. A total of 54 patients were screened and 45 received at least one dose of OPC-61815. The safety, efficacy, and pharmacodynamic analysis sets each included 45 patients. Of these, 38 patients maintained the 8 mg dose whereas 7 had a dose escalation to 16 mg (all dose increases occurred on Day 2). None of the patients who received a dose increase required a subsequent dose decrease. Forty-one patients completed the trial and 4 were discontinued (due to physician decision, n=2; TEAEs, n=1; or non-medical reasons, n=1). Early study completion was recorded for 34 patients, of whom 30 (88.2%) became capable of oral intake and 4 (11.8%) had all congestive findings resolve, requiring no further improvement of volume overload. Eighteen of the 30 patients who completed the study early because they became capable of oral intake went on to receive oral tolvaptan before the termination of the follow-up period. The total mean study duration for all patients in the safety analysis set was 2.4 days (median, 2.0 days).

Baseline patient demographic and disease characteristics are shown in
Table 1. Just over half the patients were male (51.1%); the mean (standard deviation [SD]) age and body weight were 73.7 (11.1) years and 64.5 (18.1) kg, respectively. The most frequent NYHA classification was Class IV (64.4%) followed by Class III (26.7%). Moderate or severe edema in the lower extremities was reported in a respective 26.7% and 13.3% of patients at baseline, and moderate and severe pulmonary congestion was reported in a respective 51.1% and 28.9% of patients. Pulmonary rales were reported in 80.0% of patients, third cardiac sound in 40.0%, paroxysmal dyspnea in 64.4%, and orthopnea in 73.3%. The mean (SD) respiratory rate was 21.2 (5.0) breaths/min. Patient-assessed dyspnea was reported in 80.0% of patients. The most common reason that oral intake was judged to be difficult or impossible was ongoing non-invasive positive pressure ventilation (NPPV) therapy (57.8%), followed by risk of aspiration (31.1%), and reduced swallowing function (2.2%) (Supplementary Table 1); other reasons were reported for 6 (13.3%) patients and included dyspnea (n=3), pneumonia (n=1), risk of worsening respiratory condition (n=1), and continuous use of a reservoir mask (n=1).
Table 1.
Baseline Patient and Disease Characteristics (Safety Analysis Set)
| |
OPC-61815
(N=45) |
| Sex (male) |
23 (51.1) |
| Age (years), mean (SD) |
73.7 (11.1) |
| Body weight (kg), mean (SD) |
64.5 (18.1) |
| BMI (kg/m2), mean (SD) |
25.8 (5.7) |
| NYHA class |
| Class I |
0 (0) |
| Class II |
2 (4.4) |
| Class III |
12 (26.7) |
| Class IV |
29 (64.4) |
| Unknown |
2 (4.4) |
| Lower limb edema |
| None |
7 (15.6) |
| Mild |
20 (44.4) |
| Moderate |
12 (26.7) |
| Severe |
6 (13.3) |
| Pulmonary congestion |
| None |
1 (2.2) |
| Mild |
8 (17.8) |
| Moderate |
23 (51.1) |
| Severe |
13 (28.9) |
| Jugular venous distention |
| Yes |
23 (51.1) |
| No |
21 (46.7) |
| Unknown |
1 (2.2) |
| Hepatomegaly |
| Yes |
4 (8.9) |
| No |
40 (88.9) |
| Unknown |
1 (2.2) |
| Pulmonary rales |
36 (80.0) |
| Third cardiac sound |
18 (40.0) |
| Respiratory rate (breaths/min), mean (SD) |
21.2 (5.0) |
| Paroxysmal nocturnal dyspnea |
29 (64.4) |
| Orthopnea |
33 (73.3) |
| Dyspnea status (patient-assessed) |
36 (80.0) |
| Daily urine volume, mL |
| <1,500 |
6 (13.3) |
| ≥1,500 |
15 (33.3) |
| Unknown |
24 (53.3) |
| Creatinine, mg/dL |
| <2 |
40 (88.9) |
| ≥2 |
5 (11.1) |
| Unknown |
0 (0) |
| AVP, ng/L |
| ≤3.1 |
20 (44.4) |
| >3.1 |
25 (55.6) |
| Albumin, g/dL |
| <3 |
21 (46.7) |
| ≥3 |
24 (53.3) |
| Treated with intravenous inotropes |
5 (11.1) |
Data are shown as n (%) unless otherwise indicated. AVP, arginine vasopressin; BMI, body mass index; NYHA, New York Heart Association; SD, standard deviation.
Safety data are summarized in
Table 2. Most TEAEs were mild or moderate in severity. The most frequently reported TEAE was constipation in 12 (26.7%) patients and the most frequently reported treatment-related TEAE was dry mouth in 2 (4.4%) patients. Serious TEAEs were reported in 2 (4.4%) patients (ventricular tachycardia and pleural effusion in 1 patient each). Both serious TEAEs occurred in the 8-mg group. Mild hypernatremia, a TEAE of interest, was reported in 1 (2.2%) patient (8-mg group); a relationship to OPC-61815 could not be ruled out. There were no deaths during the study, and no clinically relevant changes from baseline were found in laboratory parameters, electrocardiogram parameters, or vital signs.
Table 2.
Summary of Treatment-Emergent Adverse Events (Safety Analysis Set)
| |
OPC-61815
8 mg (n=38) |
OPC-61815
16 mg (n=7) |
Total
(N=45) |
| Patients treated |
38 (100.0) |
7 (100.0) |
45 (100.0) |
| TEAEs |
29 (76.3) |
6 (85.7) |
35 (77.8) |
| Events, n |
77 |
15 |
92 |
| Treatment-related TEAEs |
5 (13.2) |
1 (14.3) |
6 (13.3) |
| Deaths |
0 (0) |
0 (0) |
0 (0) |
| Serious TEAEs |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Treatment-related serious TEAEs |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Severe TEAEs |
2 (5.3) |
1 (14.3) |
3 (6.7) |
| Treatment-related severe TEAEs |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Discontinuation due to TEAEs |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Discontinuation due to treatment-related TEAEs |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| TEAEs occurring in ≥2 patients |
| Cardiac disorders |
| Ventricular tachycardia |
3 (7.9) |
0 (0.0) |
3 (6.7) |
| Gastrointestinal disorders |
| Constipation |
11 (28.9) |
1 (14.3) |
12 (26.7) |
| Dry mouth |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Hematochezia |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| General disorders and administration site conditions |
| Injection site inflammation |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Hepatobiliary disorders |
| Hepatic function abnormal |
5 (13.2) |
0 (0.0) |
5 (11.1) |
| Infections and infestations |
| Urinary tract infections |
4 (10.5) |
0 (0.0) |
4 (8.9) |
| Metabolism and nutrition disorders |
| Hypokalemia |
2 (5.3) |
2 (28.6) |
4 (8.9) |
| Musculoskeletal and connective tissue disorders |
| Back pain |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Psychiatric disorders |
| Delirium |
2 (5.3) |
1 (14.3) |
3 (6.7) |
| Insomnia |
4 (10.5) |
0 (0.0) |
4 (8.9) |
| Skin and subcutaneous tissue disorders |
| Dry skin |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Eczema |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Vascular disorders |
| Phlebitis |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Treatment-related TEAEs |
| Cardiac disorders |
| Ventricular tachycardia |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Gastrointestinal disorders |
| Dry mouth |
2 (5.3) |
0 (0.0) |
2 (4.4) |
| Nausea |
0 (0.0) |
1 (14.3) |
1 (2.2) |
| Metabolism and nutrition disorders |
| Hypercalcemia |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Hypernatremia |
1 (2.6) |
0 (0.0) |
1 (2.2) |
| Renal and urinary disorders |
| Renal impairment |
1 (2.6) |
0 (0.0) |
1 (2.2) |
Data are shown as n (%) unless otherwise indicated. TEAE, treatment-emergent adverse event.
The mean change in body weight from baseline decreased throughout the study (Days 2–6) and was reported as −3.01 kg on the day after final OPC-61815 administration (Figure 2A). By dose, the change in body weight at final administration was −3.14 kg for the 8-mg group and −2.20 kg for the 16-mg group (Figure 2B). Jugular venous distension decreased by 4.06 cm among the 19 patients who had a value at baseline (baseline 5.29 cm; day after final OPC-61815 administration 1.23 cm).

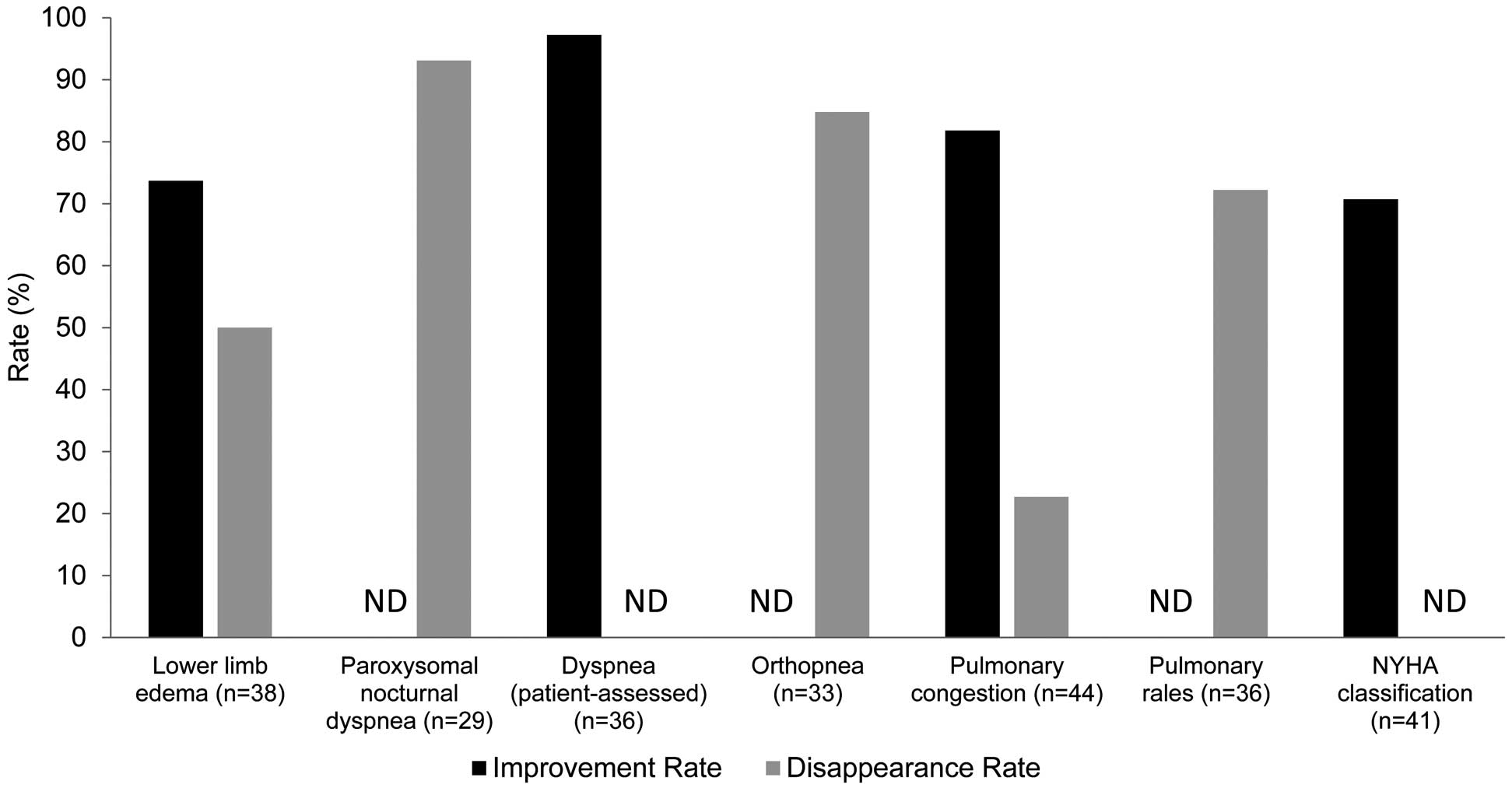
Improvement and disappearance rates for congestive and dyspnea symptoms are shown in
Figure 3. Among the 38 patients who had lower limb edema at baseline, the improvement rate was 73.7% and the disappearance rate was 50.0%. The improvement rate and disappearance rate among the 44 patients who had pulmonary congestion at baseline was 81.8% and 22.7%, respectively. The respective disappearance rates of pulmonary rales (n=36), cardiac third sound (n=18), paroxysmal nocturnal dyspnea (n=29), and orthopnea (n=33) were 72.2%, 72.2%, 93.1%, and 84.8%. The improvement rate of patient-assessed dyspnea among the 36 patients for whom it was present at baseline was 97.2%. Improvement in NYHA classification by 1 or more classes at the time of final OPC-61815 administration (vs. baseline) occurred in 70.7% of patients. One patient improved by 3 classes, 12 improved by 2 classes, and 16 improved by 1 class. Results for all congestive and dyspnea symptoms, and NYHA classification were similar between patients in the 8-mg and 16-mg dose groups.
Daily urine volume increased from Day 1 (baseline) on Day 2 and Day 3 following OPC-61815 treatment (Figure 4A). The mean (SD) change from Day 1 (baseline) for both groups combined was 581.5 (989.8) mL on Day 2 and 414.6 (1,651.7) mL on Day 3 (Figure 4B). In the 8-mg (n=15) and 16-mg (n=6) groups, the change in daily urine volume from Day 1 (baseline) to Day 2 was 888.5 mL and −185.8 mL, respectively. During the first day of OPC-61815 treatment, urine volume (mL/h) started to increase at 0- to 1-h post-dose and reached a maximum level at 1–2 h post-dose (Figure 4C). Three patients had a daily urine output of ≥5,000 mL, and TEAEs reported in these patients included hypernatremia, hypokalemia, ventricular tachycardia, and increased blood pressure. There were no TEAE-related withdrawals in these patients; however, a causal relationship with OPC-61815 could not be ruled out for the case of mild hypernatremia.

During the treatment period, there was a trend for increased concentrations in both serum sodium and serum potassium (Figure 5A,5B) and a trend for increased serum osmolality (Figure 5C). There were no obvious fluctuations in blood pressure and heart rate during the treatment period (Figure 5D,5E). Furthermore, there were no obvious differences in blood pressure or heart rate between patients who were administered intravenous inotropes and those who were not (data not shown). During the treatment period, significant increases were observed in plasma AVP and plasma renin activity on the day after final OPC-61815 administration compared to baseline, and significant decreases were observed in plasma BNP, serum NT-proBNP, and serum troponin I concentrations (all P<0.05) (Table 3). There were no significant changes in serum creatinine and blood urea nitrogen levels from baseline.

Table 3.
Change in Biomarker Concentrations (Pharmacodynamic Analysis Set)
| |
Baseline
(n=45) |
Day after final OPC-61815
administration (n=45) |
P value |
| Serum creatinine, mean (SD) (mg/dL) |
1.14±0.56 |
1.10±0.53 |
0.071 |
| BUN, mean (SD) (mg/dL) |
21.9±10.1 |
20.7±9.2 |
0.199 |
| Plasma AVP (pg/mL) |
3.5 (2.0, 5.0) |
5.3 (3.6, 8.5) |
<0.001* |
| Plasma renin activity (ng/mL/h) |
0.7 (0.2, 1.4) |
0.8 (0.4, 2.1) |
0.036* |
| Plasma BNP (pg/mL) |
292.0 (114.0, 543.0) |
190.0 (79.9, 474.0) |
<0.001* |
| Serum NT-proBNP (ng/L) |
3,340 (1,210, 7,290) |
2,440 (736, 4,460) |
<0.001* |
| Serum troponin I (ng/L) |
35.2 (20.3, 121.0) |
30.9 (14.4, 59.4) |
<0.001* |
Data are shown as median (interquartile range), unless otherwise indicated. AVP, arginine vasopressin; BNP, B-type natriuretic peptide; BUN, blood urea nitrogen; NT-pro BNP, N-terminal proBNP; SD, standard deviation. For non-normal distributions (plasma AVP, plasma renin activity, plasma BNP, serum NT-proBNP, and serum troponin I), paired t-tests were performed after log transformation. *Significant difference (P<0.05; paired t-test) between the day after final OPC-61815 administration and baseline.
The mean (SD) duration of exposure was 2.4 (1.4) days for all treated patients and 2.3 (1.4) or 2.9 (1.2) days for patients who maintained the 8-mg dose or those who had their dose increased to 16 mg, respectively (Supplementary Table 2). The daily duration of exposure for Days 1–5 is also shown in
Supplementary Table 2.
Discussion
This open-label study was designed to evaluate the tolerability of OPC-61815 in CHF patients who have difficulty with, or an inability to, take oral medications. OPC-61815 was assessed in a recent Phase III non-inferiority validation study (ClinicalTrials.gov [NCT03962101]), which used tolvaptan tablets as a comparator. However, patients who had difficulty with oral intake were excluded from that study, necessitating an additional clinical analysis of OPC-61815 in this patient population. Our data confirmed the tolerability of once daily (up to 5 days) intravenous OPC-61815 (8 or 16 mg) in patients with CHF who were incapable of oral intake. Of the 45 patients who were treated with OPC-61815, 41 (91.1%) patients completed the study; 38 (84.4%) patients maintained an 8-mg dose, whereas 7 (15.6%) patients had a dose increase to 16 mg. NPPV use was the most common reason for difficulty with oral intake (n=26 [57.8%]).
When using aquaretic agents to treat patients who have difficulty with oral intake, extra care should be taken to ensure rapid correction of hyponatremia, hypernatremia, and dehydration. In addition, the administration of OPC-61815 may result in excessive diuresis. Thus, when OPC-61815 is administered to such patients, it is critical to monitor the fluid balance and to measure serum electrolyte concentrations. To address this, the fluid balance was checked hourly until 2 h after the start of treatment, every 2 h until 8 h, and then daily during the treatment period. Additionally, serum electrolyte concentrations (Na, K) were measured at 4 h and 8 h after treatment initiation and daily thereafter. It should also be noted that because the target population of the present study was patients for whom oral intake was difficult or impossible, the starting dose of OPC-61815 was 8 mg, which is half the maximum dose. In this study, 1 (2.2%) patient had a TEAE of hypernatremia, which was mild in severity. This is a lower incidence than is reported in postmarketing studies of tolvaptan tablets (4.4%),21
but is similar to the incidence reported in the Phase III non-inferiority study (2.7%, ClinicalTrials.gov [NCT03962101]). No patients in our study had an TEAE of serum sodium concentration ≥150 mEq/L and there were no incidences of serum sodium concentrations that showed an increase of >10 mEq/L within 24 h of OPC-61815 administration. One patient was discontinued due to a ventricular tachycardia on Day 1. This patient had been administered 8 mg OPC-61815 on Day 1 of the study and developed multifocal ventricular tachycardia 4 h after administration, which spontaneously stopped approximately 10–15 s later. Ventricular tachycardia developed again on the same day. The study investigator reported that, although OPC-61815 had increased urine output, it was likely that the intravenous bolus administration of 20 mg furosemide 30 min prior caused the rapid increase in urine output, thus directly causing the ventricular tachycardia. As such, this event was assessed as a treatment-related severe TEAE.
Regarding dosing, no safety risks specific to an increased dose of 16 mg were identified; safety outcomes were similar regardless of dose, and no deaths were reported. Overall, this study confirmed earlier publications that found that there are no serious or significant safety risks associated with OPC-61815 treatment (ClinicalTrials.gov [NCT03962101]).21
Efficacy outcomes were comparable with the prior non-inferiority study, although some measures showed greater improvement in our analysis. Notably, body weight decreases were greater in the present study (mean −3.01 kg) compared with the prior Phase III study (maximum Day 6 −1.63 kg, ClinicalTrials.gov [NCT03962101]). The improvement rate for pulmonary congestion was also higher in the present study (81.8%) than in the Phase III study (56.1%, ClinicalTrials.gov [NCT03962101]). In this analysis, moderate and severe lower limb edema at baseline was observed in 18 (40.0%) patients, and dyspnea in 36 (80.0%). These patients experienced high rates of improvement and disappearance. There was a 73.7% improvement rate and a 50% disappearance rate for patients with lower limb edema, which was comparable to the 68.9% improvement rate observed in the Phase III study (ClinicalTrials.gov [NCT03962101]). Those with dyspnea had a 97.2% improvement rate (not reported for the Phase III study). Additionally, the disappearance rate was 93.1% for paroxysmal nocturnal dyspnea, 84.8% for orthopnea, and 72.2% for pulmonary rales (not reported for the Phase III study). Finally, the present study had a higher improvement rate for NYHA classification (70.7%) compared with the Phase III non-inferiority study (44.9%, ClinicalTrials.gov [NCT03962101]). There are several things to consider when comparing the results of these 2 studies. First, when noting the NYHA classification and congestive findings at baseline, there was a larger percentage of patients in the acute exacerbation stage of HF in the present study than in the Phase III study. At baseline, 64.4% (29/45) and 26.7% (12/45) of patients in the present study had Class IV or Class III HF, respectively; these proportions were much higher than that observed for the participants of the Phase III non-inferiority study (Class IV n=2 [0.7%]; Class III n=60 [20.4%], ClinicalTrials.gov [NCT03962101]). There was also a higher retention of body fluid prior to administration of OPC-61815 in the present study. Finally, the differences between a confirmatory study and a tolerance study must be considered. For example, in the Phase III study, patients were only included if their weight was relatively stable (ClinicalTrials.gov [NCT03962101]), whereas in the present tolerability study, patients with greater fluctuations in body weight were allowed. Additionally, changes in diuretic dose and the use of an infusion solution were permitted in this tolerability study.
This is the first publication to report the hourly diuretic effect of OPC-61815. Compared with baseline, the urine excretion rate (hourly urine volume) began to increase 0–1 h after the start of OPC-61815 administration and maximum urine excretion rate was observed between 1 and 2 h. In the Phase II study, the Tmax
for urine excretion was 1.76 h and 1.52 h in the OPC-61815 8-mg and 16-mg groups, respectively,22
which coincides with the time of maximum urine excretion rate reported in the present study. For tolvaptan, the Tmax
reported in the Phase II study was 4.07 h.22
Taken together, these data suggest that OPC-61815 may produce an aquaretic effect earlier than oral tolvaptan. However, because the protocol for the present study did not define fixed dosages for concomitant diuretics (i.e., changes in dose were allowed), their effects cannot be excluded. It should also be noted that baseline and hourly urine excretion rate was only obtained from about half of the enrolled patients (n=21) and the baseline rate was calculated dividing the 24-h urine output by 24.
In our study, after OPC-61815 administration, plasma AVP and plasma renin activity significantly increased, and plasma BNP significantly decreased (Table 3). The increase in plasma renin activity may be caused by hypovolemia owing to the diuretic effect of OPC-61815. No clear change was observed in renal function and there are no obvious differences when compared with tolvaptan in the QUEST study.15
In fact, the Phase III non-inferiority study (ClinicalTrials.gov [NCT03962101]) that compared OPC-61815 and tolvaptan showed no obvious difference in the changes in these parameters.
Study Limitations
This study had several limitations. First, only Japanese patients were included, thus limiting the generalizability of the study findings. Second, few patients met the dose increase criteria and had their dose increased to 16 mg; therefore, findings for the 16-mg group should be interpreted with caution. However, there were no notable differences in tolerability or any study outcomes between the 2 dose groups. Third, there was no comparator group. Fourth, changes in the dose of concomitant diuretics were allowed. Finally, because there were differences in both the inclusion/exclusion criteria and the study design between the present study and previous studies of OPC-61815 and tolvaptan, the comparison of study endpoints is limited.
Conclusions
In this assessment of intravenous OPC-61815 treatment in patients for whom oral intake was difficult or impossible, we found that the study drug was well tolerated, and the previously reported safety and efficacy of OPC-61815 were confirmed in this patient population. Together, these findings indicate that OPC-61815 may be considered for patients who are unable to ingest oral aquaretic treatment.
Acknowledgments
The authors thank Sarah Bubeck, PhD, of Edanz (http://www.edanz.com) for providing medical writing support, which was funded by Otsuka Pharmaceutical Co. Ltd., in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Sources of Funding
This study was financially supported by Otsuka Pharmaceutical Co., Ltd.
Disclosures
K.K. has received lecture fees from Otsuka Pharmaceutical Co., Ltd. and is a member of the
Circulation Journal’s Editorial Team; E.N., T.H., and S.K. are employees of Otsuka Pharmaceutical Co., Ltd.
IRB Information
Review and approval of the study protocol and associated documentation was provided by the Institutional Review Board or Independent Ethics Committee at each of the 30 participating study sites. The representative Institutional Review Board was at the University of Toyama (#2019-05-A001).
Data Availability
The deidentified participant data, data dictionaries, study protocol, and statistical analysis plan will be shared on a request basis, upon provision of a methodologically sound meta-analysis proposal. Data will be available after marketing approval in global markets, or starting 1–3 years following publication. There is no end date to the availability of the data. Please contact the corresponding author directly to request data sharing.
Contributor Statements
(1) Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work: K.K., E.N., T.H., S.K.
(2) Drafting the work or revising it critically for important intellectual content: K.K., E.N., T.H., S.K.
(3) Final approval of the version to be published: K.K., E.N., T.H., S.K.
(4) Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: K.K., E.N., T.H., S.K.
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-21-0926
References
- 1.
GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018; 392: 1789–1858.
- 2.
Konishi M, Ishida J, Springer J, von Haehling S, Akashi YJ, Shimokawa H, et al. Heart failure epidemiology and novel treatments in Japan: Facts and numbers. ESC Heart Fail 2016; 3: 145–151.
- 3.
Shimokawa H, Miura M, Nochioka K, Sakata Y. Heart failure as a general pandemic in Asia. Eur J Heart Fail 2015; 17: 884–892.
- 4.
O’Connor CM, Stough WG, Gallup DS, Hasselblad V, Gheorghiade M. Demographics, clinical characteristics, and outcomes of patients hospitalized for decompensated heart failure: Observations from the IMPACT-HF registry. J Card Fail 2005; 11: 200–205.
- 5.
Gheorghiade M, Follath F, Ponikowski P, Barsuk JH, Blair JE, Cleland JG, et al. Assessing and grading congestion in acute heart failure: A scientific statement from the acute heart failure committee of the Heart Failure Association of the European Society of Cardiology and endorsed by the European Society of Intensive Care Medicine. Eur J Heart Fail 2010; 12: 423–433.
- 6.
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18: 891–975.
- 7.
Tsutsui H, Isobe M, Ito H, Ito H, Okumura K, Ono M, et al. JCS 2017/JHFS 2017 Guideline on diagnosis and treatment of acute and chronic heart failure: Digest version. Circ J 2019; 83: 2084–2184.
- 8.
Palazzuoli A, Ruocco G, Pellegrini M, Beltrami M, Del Castillo G, Nuti R. Loop diuretics strategies in acute heart failure: From clinical trials to practical application. Curr Drug Targets 2015; 16: 1246–1253.
- 9.
Mentz RJ, Kjeldsen K, Rossi GP, Voors AA, Cleland JG, Anker SD, et al. Decongestion in acute heart failure. Eur J Heart Fail 2014; 16: 471–482.
- 10.
Gottlieb SS, Brater DC, Thomas I, Havranek E, Bourge R, Goldman S, et al. BG9719 (CVT-124), an A1 adenosine receptor antagonist, protects against the decline in renal function observed with diuretic therapy. Circulation 2002; 105: 1348–1353.
- 11.
Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011; 364: 797–805.
- 12.
Yamamura Y, Nakamura S, Itoh S, Hirano T, Onogawa T, Yamashita T, et al. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: Pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J Pharmacol Exp Ther 1998; 287: 860–867.
- 13.
Udelson JE, Bilsker M, Hauptman PJ, Sequeira R, Thomas I, O’Brien T, et al. A multicenter, randomized, double-blind, placebo-controlled study of tolvaptan monotherapy compared to furosemide and the combination of tolvaptan and furosemide in patients with heart failure and systolic dysfunction. J Card Fail 2011; 17: 973–981.
- 14.
Matsue Y, Ter Maaten JM, Suzuki M, Torii S, Yamaguchi S, Fukamizu S, et al. Early treatment with tolvaptan improves diuretic response in acute heart failure with renal dysfunction. Clin Res Cardiol 2017; 106: 802–812.
- 15.
Matsuzaki M, Hori M, Izumi T, Fukunami M, Tolvaptan Investigators. Efficacy and safety of tolvaptan in heart failure patients with volume overload despite the standard treatment with conventional diuretics: A phase III, randomized, double-blind, placebo-controlled study (QUEST study). Cardiovasc Drugs Ther 2011; 25(Suppl 1): S33–S45.
- 16.
Kuragaichi T, Sato Y. Temporal trends of a vasopressin V2 receptor antagonist in heart failure using a nationwide database in Japan. ESC Heart Fail 2021; 8: 527–538.
- 17.
Otsuka Pharmaceutical Co., Ltd. Jynarque: Highlights of prescribing information. https://www.otsuka-us.com/media/static/JYNARQUE-PI.pdf (accessed June 5, 2021).
- 18.
Otsuka Pharmaceutical Co., Ltd. Samsca: Highlights of prescribing information. https://www.otsuka-us.com/media/static/Samsca-PI.pdf (accessed June 5, 2021).
- 19.
Center for Drug Evaluation and Research. Jynarque: Clinical pharmacology review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/204441Orig1s000ClinPharmR.pdf (accessed June 5, 2021).
- 20.
Sica DA. Diuretic-related side effects: Development and treatment. J Clin Hypertens (Greenwich) 2004; 6: 532–540.
- 21.
Kinugawa K, Sato N, Inomata T, Yasuda M, Shimakawa T, Fukuta Y. Real-world effectiveness and tolerability of tolvaptan in patients with heart failure: Final results of the Samsca post-marketing surveillance in heart failure (SMILE) study. Circ J 2019; 83: 1520–1527.
- 22.
Sato N, Uno S, Yamasaki Y, Hirano T, Kim S on behalf of the OPC-61815 Investigators. Pharmacokinetics, pharmacodynamics, efficacy, and safety of OPC-61815, a prodrug of tolvaptan for intravenous administration, in patients with congestive heart failure: A Phase II, Multicenter, Double-Blind, Randomized, Active-Controlled Trial. Circ J, doi:10.1253/circj.CJ-21-0430.
