Aging is a physiological process that is associated with the enhancement of unfavorable mechanisms. Lopez-Otin et al1 recently summarized 12 hallmarks of aging: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, disabled macroautophagy, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis. The biological process of aging is complex and does not allow a simple approach to comprehensive understanding. My group has shown the central pathogenic role of cellular senescence in cardiovascular metabolic disorders.2–13 Eukaryotic cells have a limited capacity for proliferation, eventually stop dividing. When cells are exposed to toxic stress, including reactive oxygen species (ROS), they also stop proliferating. These processes are respectively described as replicative and premature senescence.1 Senescent cells become anti-apoptotic, show alterations in the genetic expression profile, and acquire a pro-inflammatory phenotype known as the senescence-associated secretory phenotype (SASP).1,11 In vitro studies have revealed aging as contagious, causing young cells to exhibit a senescent phenotype through a bystander effect.14 “Irreversible termination of proliferation” is the classical definition of cellular senescence, and this is well accepted as an antitumorigenic mechanism contributing to the inhibition of the uncontrollable proliferation of cells that leads to tumorigenesis. “Senolysis” (i.e., the specific depletion of senescent cells) has been shown to reverse aging in genetic/pharmacologic/vaccination models.2,15–17 Compounds that mediate senolysis are termed “senolytics” and of them, dasatinib and quercetin (D+Q) are being extensively studied.12,18–20 Studies using rodents showed that D+Q reversed aging, and these are now being tested in clinical trials.21
One of the mysterious components of aging is the synchronization of this biological process. Some tissues are highly proliferative, whereas others are mostly terminally differentiated. However, they show functional decline in parallel with aging. With chronological aging or with heart failure, some hormones, proteins, vitamins, and microRNAs are known to increase or reduce in circulation, mediating pro- or anti-aging effects.22,23 Molecules, including metabolites, may have roles in “sync-aging”, so exploration of prosenescent proteins (senoproteins) or metabolites (senometabolites) is an important approach for aging research.
Main Areas of Research
p53 Signaling Mediates Cellular Senescence
The p53-signaling pathway coordinates DNA repair, apoptosis, and senescence to preserve genomic stability and prevent tumorigenesis.11 Several types of cancers are known to have a loss-of-function mutation for p53, and for this reason, this protein is described as a “guardian of the genome”. Besides its protective function, p53 is known to have a bidirectional role.11 Telomere attrition, a wide variety of stress signals, including DNA damage, oxidative stress, and oncogene activation, increase p53 signaling and mediate cellular senescence.11 Senescent cells show both irreversible termination of proliferation and the SASP phenotype,1,11 which promotes functional and structural remodeling in organs, making the organism prone to developing age-related disorders.
Pathogenic Role of p53 Signaling in Cardiovascular-Metabolic Diseases
Activation of p53 has been observed in aged vessels, and failing hearts, and is recognized as promoting pathogenesis in atherosclerosis or heart failure.9,10 p53 was increased in the visceral fat from patients with type 2 diabetes, and in rodents p53 induced chronic sterile inflammation in visceral adipose tissue and promoted systemic glucose intolerance.7 Semaphorin3E (sema3E) is a secreted-type semaphorin, and plexinD1 is its receptor. The sema3E/plexinD1 axis is reported to have roles in embryogenesis, but its role in the adult stage is unknown. My group found that p53-mediated signaling increased the production of sema3E in visceral fat, which led to increased infiltration of plexinD1-positive inflammatory macrophages into the fat tissue and introduced chronic sterile inflammation and systemic insulin resistance (I/R).6 Peptide vaccination therapy targeting sema3E inhibited this protein and ameliorated systemic I/R under conditions of dietary obesity.3 Systemic I/R, a state of hyperinsulinemia, is also known to develop in heart failure. Clinical trials indicate diabetic drugs that ameliorate hyperinsulinemia and do not signal through insulin reduce the risk of heart failure.24 The link between hyperinsulinemia and heart failure is unclear. My group previously showed that p53-mediated signaling promoted chronic sterile inflammation in visceral fat, which induced systemic I/R in a mouse model of left ventricular (LV) pressure overload.13 We also found the level of I/R varied among organs; for example, visceral fat acquired I/R in the chronic phase of LV pressure overload, but I/R was continuously activated in cardiac tissue, inducing excessive cardiomyocyte hypertrophy, tissue hypoxia, and reduced cardiac function.13,25 Mechanistically, continuous activation of adrenergic signaling induced lipolysis, which caused production of ROS and an increase in the p53 level.13 Sympathetic nerve activation develops with heart failure, which is well known to associate with poor prognosis. Activation of sympathetic nerve signaling increases the p53 levels in endothelial cells and bone marrow cells, which leads to an increase in endothelial cells’ Icam1 and integrin levels in bone marrow-derived cells and enhances chronic inflammation in the heart under LV pressure overload.5 Capillary rarefaction develops in the failing heart under LV pressure overload.26 Catecholamine increases the ROS levels in endothelial cells and induces apoptosis in these cells, enhancing the capillary rarefaction.27 A Sglt2 inhibitor, empagliflozin, ameliorated this process, enhanced capillarization, and improved systolic cardiac dysfunction.27 The level of endothelial cells’ p53 also increases under hyperglycemia, which reduces endothelial dysfunction.4 These findings indicate that inhibition of p53 signaling can suppress pathologies in cardiovascular-metabolic diseases.
Senolysis as a Strategy to Reverse Aging Phenotype
Suppression of cellular senescence increases the risk of tumorigenesis, and safer approaches to suppressing the accumulation of senescent cells need to be explored. Recently, genetic as well as pharmacologic approaches showed that specific depletion of senescent cells reversed the aging phenotype in mice.15–17 Genetic models designed to deplete p16-positive cells showed a reversal of the aging phenotype and reduced atherosclerosis.15 D+Q and ABT263 are the most extensively studied senolytics in both in vitro and in vivo experiments.17 The combination D+Q is now being tested in patients with chronic kidney disease21 or lung fibrosis.28 Preliminary results showed a reduction of senescent markers in subcutaneous adipose tissue.21 Many of the currently identified senolytics are classified as anticancer drugs,12 so there are potential safety issues. Finding less toxic senolytics continues to be an interesting research field. Recently, the level of glycoprotein nonmetastatic melanoma protein B (Gpnmb) at the cell membrane was found to increase with aging,2,29 and vaccination therapy designed to eliminate Gpnmb-positive cells reduced the number of p19-positive cells, reversed atherosclerosis, and improved systemic glucose intolerance.2
Brown Adipose Tissue (BAT) as an Interesting Target of Aging Research
White adipose tissue has roles in the storage of lipids and the production of adipokines, whereas BAT was initially characterized as an organ involved in thermogenesis. It was once considered to be abundant only in newborn humans and small rodents, but in 2009, a PET-CT scan showed that human adults also possess active BAT.30 Interestingly, detectable BAT decreases with obesity and aging.30 In addition to its thermogenic function, studies indicate that BAT contributes to systemic metabolism due to its high oxidative capacity.31–37 BAT is a highly vascularized organ with abundant mitochondria. It has a high capacity to uptake glucose and fatty acids and dissipate these substrates for heat emission. A high-calorie diet reduces the level of vascular endothelial growth factor-A (VEGF-A) and induces capillary rarefaction in BAT.32 Hypoxic conditions in BAT increase the level of hypoxia-inducible factor 1-alpha (HIF-1α) in this organ. HIF-1α is reported as not angiogenic in white adipose tissue,38 and does not increase VEGF-A or promote an angiogenic response in BAT.32 Instead, it enhanced continuous activation of mitophagy and reduced the number of mitochondria in brown adipocytes, which led to a “whitened phenotype” of BAT characterized by a functional decline that resulted in reduced glucose tolerance.32 Metabolic stress causes BAT dysfunction, but the underlying mechanisms are unclear. Dietary obesity leads to a marked increase in tissue factor (TF), coagulation factor VII, and FXa in BAT.33 The TF-FVIIa (activated form of FVII)-FXa complex is known to activate proteinase-activated receptor1 (PAR1), and there was a significant increase in PAR1 expression in BAT upon metabolic stress in a murine dietary obese model, from which my group concluded that FXa-PAR1 signaling promotes BAT dysfunction and systemic metabolic disorder.33 Mass spectrometric analysis showed a significant increase in gamma-aminobutyric acid (GABA) levels in the BAT of dietary obese mice, and the expression of GABA-B receptor1 (GABA-BR1) significantly increased in the cell membrane fraction of BAT of these mice.32 Signaling mediated by GABA-GABA-BR1 contributes to the development of systemic metabolic dysfunction through inhibition of BAT function in obesity.32 As already noted, BAT is a highly vascularized organ, so enhancement of angiogenesis is one approach, and maintaining capillarization is another approach. In BAT, a high-calorie diet induces apoptosis in endothelial cells, and polyphenol suppressed this via a SIRT-1 dependent pathway, contributing to the maintenance of the capillary network and thermogenic response in BAT.34 Low body temperature predicts poor prognosis in patients with heart failure, but the underlying mechanisms and pathologic implications are still largely unknown. My group found that BAT dysfunction developed in a murine thoracic aortic constriction heart failure model and had a causal role in promoting pathologies in the failing heart.39 Metabolomic analyses showed that BAT dysfunction led to an increase in trimethylamine N-oxide (TMAO), and administration of TMAO suppressed cardiac function during LV pressure overload.39 Mechanistically, TMAO induced mitochondrial damage by inhibiting cytochrome c oxidase 1 (COX1), a chief component of mitochondrial complex IV.39 These studies indicate that maintenance of BAT homeostasis is critical to inhibit progression of the pathogenesis of cardiovascular–metabolic diseases.
“Think-Aging” Through the Concept of “Sync-Aging”
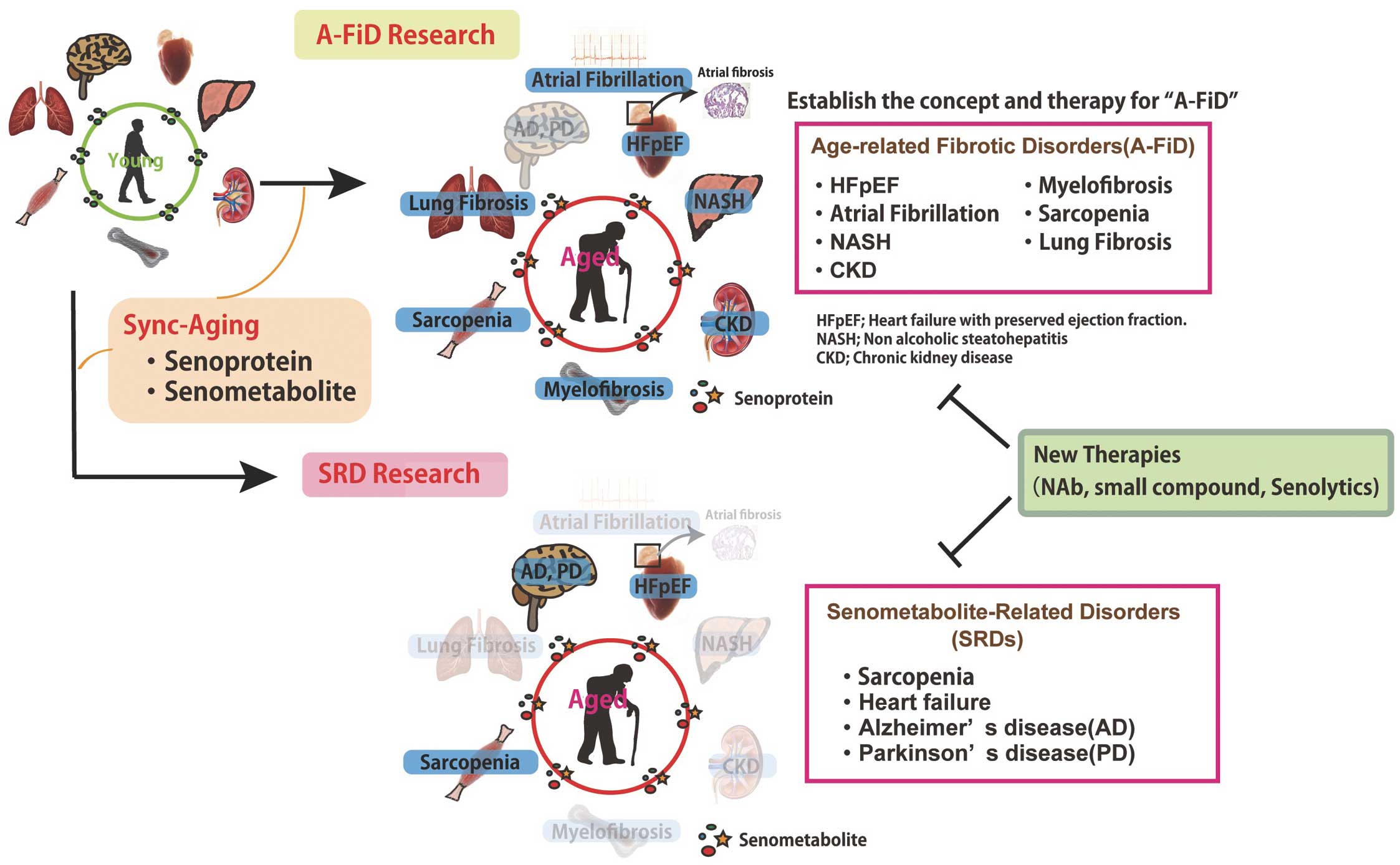
Studying cellular senescence and metabolically active organs including BAT, led to consideration of the synchronization of aging (sync-aging) as interesting and mysterious. Proliferative capacity and level of differentiation vary among organs, but they all seem to show a functional decline in parallel with aging. Mechanisms contributing to sync-aging are yet to be defined, but some hormones, proteins, vitamins, and microRNAs are reported to increase or reduce in circulation, mediating pro- or anti-aging effects.22 Molecules such as proteins and metabolites may have roles in sync-aging, so here I define “senometabolites” as circulating metabolites with causal roles in the synchronization and progression of aging. I also define “senometabolite-related diseases” (SRDs) as age-related disorders that develop through mitochondrial dysfunction. SRDs include heart failure, sarcopenia, Alzheimer’s disease, and Parkinson’s disease, and can be considered as one syndrome sharing similar pathogenesis through senometabolites. Focusing on senoprotein, I also categorize heart failure with preserved ejection fraction (HFpEF), atrial fibrillation, sarcopenia, chronic kidney disease (CKD), and nonalcoholic steatohepatitis (NASH) as one syndrome defined as “age-related fibrotic disorders” (A-FiDs). Catheter ablation can be an effective treatment of atrial fibrillation, although recurrence continues to be a problem. Therapies for other A-FiDs are extremely limited, simply because we do not yet know the key pathogenic mechanisms. The global population of affected patients is considered to high: HFpEF (13 million), NASH (3–5%), CKD (10–12%). It is urgent to establish next-generation therapies and by comprehensively defining them as A-FiD, we can direct research towards achieving this goal.
Acknowledgments
I thank Professor Tohru Minamino, Professor Issei Komuro, Professor Kenneth Walsh, all my colleagues and friends, and family members for their continuous mentorship and support. This work was supported by Fusion Oriented Research for disruptive Science and Technology (JST FOREST Program) (JPMJFR200L), JSPS Grants-in-Aid for Scientific Researcher (B), AMED Project for Elucidating and Controlling Mechanisms of Aging and Longevity under Grant Number JP21gm5010002, JSPS KAKENHI, Grant-in-Aid for Challenging Exploratory Research (17K19648), Grants-in-Aid for Encouragement of Young Scientists (A) (16H06244), JSPS KAKENHI, Grant-in-Aid for Challenging Exploratory Research 19K22616, Grants-in-Aid for Young Scientists (Start-up) (KAKENHI) (Research Project Number: 26893080), Intramural Research Fund for Cardiovascular Diseases of the National Cerebral and Cardiovascular Center, Daiichi Sankyo’s research grant (TaNeDS), Foundation for Medical & Pharmaceutical Research, the Uehara Memorial Foundation, Kowa Life Science Foundation, Manpei Suzuki Diabetes Foundation, MSD Life Science Foundation, Public Interest Incorporated Foundation, Kanae Foundation Research Grant, Inamori Foundation, TERUMO FOUNDATION for LIFE SCIENCES and ARTS, SENSHIN Medical Research Foundation, Takeda Science Foundation, ONO Medical Research Foundation, The Nakajima Foundation, Japan Heart Foundation Research Grant, The Senri Life Science Foundation, SUZUKEN memorial foundation, HOKUTO Corporation, Mochida Memorial Foundation for Medical & Pharmaceutical Research, The Cell Science Research Foundation, Daiichi Sankyo Foundation of Life Science, Tokyo Biochemical research Foundation, The Nakatomi Foundation, Astellas Foundation for Research on Metabolic Disorders, Grant From Japan Cardiovascular Research Foundation, Kimura Memorial Heart Foundation, Japan Diabetes Foundation, Research grant from Naito Foundation, Grant for Basic Science Research Projects from The Sumitomo Foundation, Tsukada grant for Niigata University Medical Research, The Japan Geriatrics Society, Japan Diabetes Foundation and Novo Nordisk Pharma Ltd., Kobayashi Magobei Memorial Medical Promotion Foundation of Japan, President’s Grant for Interfaculty Collaboration, Juntendo University, Japan Foundation for Applied Enzymology, Japanese Circulation Society Grant for Future-Pioneering Doctors for Basic Research.