Abstract
Background: Foam cell formation is an important step for atherosclerosis (AS) progression. We investigated the mechanism by which the long non-coding RNA (lncRNA) nuclear-enriched abundant transcript 1 (NEAT1) regulates foam cell formation during AS progression.
Methods and Results: An in vivo AS model was created by feeding ApoE−/−
mice a high-fat diet. Oxidized low-density lipoprotein (ox-LDL)-stimulated macrophages were used as a cellular AS model. Interactions between NEAT1, miR-17-5p, itchy E3 ubiquitin protein ligase (ITCH) and liver kinase B1 (LKB1) were analyzed. NEAT1 and ITCH were highly expressed in clinical samples collected from 10 AS patients and in ox-LDL-treated macrophages, whereas expression of both miR-17-5p and LKB1 was low. ITCH knockdown inhibited ox-LDL-induced lipid accumulation and LDL uptake in macrophages. Mechanistically speakingly, ITCH promoted LDL uptake and lipid accumulation in macrophages by mediating LKB1 ubiquitination degradation. NEAT1 knockdown reduced LDL uptake and lipid accumulation in macrophages and AS progression in vivo. NEAT1 promoted ITCH expression in macrophages by acting as a sponge for miR-17-5p. Inhibition of miR-17-5p facilitated ox-LDL-induced increase in LDL uptake and lipid accumulation in macrophages, which was reversed by NEAT1/ITCH knockdown.
Conclusions:NEAT1 accelerated foam cell formation during AS progression through the miR-17-5p/ITCH/LKB1 axis.
Atherosclerosis (AS) is a chronic inflammatory disease characterized by excessive deposition of cholesterol in artery walls, which is the pathological basis of various cardiovascular diseases.1 Currently, there are no effective treatment options for AS and no effective prevention measures.2 Therefore, there is an urgency to find novel treatment strategies for AS, and understanding the pathological mechanism of AS is key to achieving this goal. Circulating monocytes migrate to the subintima of the artery and differentiate into macrophages during AS progression.3 These cells transform into lipid-rich foam cells by phagocytosis of large amounts of oxidized low-density lipoprotein (ox-LDL) through scavenging receptors on their cell surface. Foam cells produce reactive oxygen species and induce an inflammatory response that recruits more macrophages into the vascular lining, leading to lipid accumulation and plaque formation on the vascular wall.4 Therefore, understanding the regulatory mechanism of foam cell formation during AS progression will provide new strategies for the prevention and treatment of AS.
Most of the transcription products of the human genome are non-coding RNAs.5 “Long non-coding RNAs” (lncRNAs) refers to a class of non-coding RNAs with a length of more than 200 nucleotides (nt).6 It has been widely reported that lncRNAs are involved in regulating AS progression.7 For example, Tang et al demonstrated that lncRNA zinc finger antisense 1 (ZFAS1) was significantly upregulated in ox-LDL-treated macrophage-derived foam cells, and that ZFAS1 knockdown reduced inflammation and lipid accumulation in foam cells.8 Nuclear-enriched abundant transcript 1 (NEAT1) is a lncRNA molecule transcribed by the multiple endocrine neoplasia 1 (MEN1), and is mainly distributed in the nucleus. Several studies have revealed the function of lncRNA NEAT1 in regulating AS progression. For example, Zhang et al reported that NEAT1 promoted AS development by facilitating the proliferation of ox-LDL-treated human aortic endothelial cells and reducing cell apoptosis.9 Of note, it has been shown that lncRNA NEAT1 expression in THP-1 cells, which derived from human monocytic leukemia, was significantly increased by ox-LDL stimulation and that NEAT1 knockdown impeded inflammatory responses and lipid uptake by THP-1 cells.10 All these results suggest that NEAT1 may be involved in the regulation of foam cell formation during AS progression, although the mechanism remains further investigation.
MicroRNAs (miRNAs) are non-coding RNAs with a length of approximately 22 nt that are involved in many biological processes.11 Dysregulation of miRNAs is an important inducing factor of the pathogenesis and development of AS.12 For example, it was previously reported that miR-576 upregulation inhibited ox-LDL-induced release of inflammatory factors from macrophages.13 miR-17-5p is a member of the miR-17-92 gene cluster and is a highly conserved miRNA that is highly expressed in the heart, lung, and so on. A previous study reported that miR-17-5p was downregulated in the plasma of patients with coronary artery disease (CAD),14 revealing the potential role in cardiovascular diseases. Notably, a previous study identified miR-17-5p as a circulating biomarker for the severity of coronary AS.15 More importantly, it was previously reported that miR-17-5p overexpression markedly inhibited the proliferation and inflammatory response of macrophages in AS patients.16 As is known, lncRNAs function as competing endogenous RNAs (ceRNAs) by acting as a sponge for miRNAs, in turn affecting the binding of miRNAs to their targets, which is described as a novel RNA-regulation mechanism.17 As proof, it was shown that silencing of NEAT1 suppressed the ox-LDL-induced inflammation response and lipid uptake in THP-1 cells by acting as a sponge miR-342-3p.10 In the present study, by using starBase database, we found that NEAT1 had a potential binding site with miR-17-5p. However, the relationship between NEAT1 and miR-17-5p in regulating foam cell formation during AS progression is largely unknown, and is worth further investigation.
Itchy E3 ubiquitin protein ligase (ITCH) is an E3 ubiquitin ligase with a molecular weight of 854 kDa that is responsible for binding substrates and transferring them to ubiquitin-containing E2 ubiquitin coupling enzymes.18 A recent study revealed that ITCH acted as an immunotherapeutic target in AS by modulating T cell activation.19 In addition, it has been reported that ITCH deficiency reduces hepatic lipid infiltration and delays AS progression by increasing fatty acid oxidation in apolipoprotein E-deficient (ApoE−/−) mice.20 In the current research, it was predicted that ITCH could ubiquitinate liver kinase B1 (LKB1) through Ubibrowser database. It has been reported that LKB1 expression is markedly reduced in human plaques, and its knockdown in macrophages accelerated foam cell formation and the AS process.21 Based on the above evidence, we put forward a hypothesis that NEAT1 promotes the uptake of lipids by macrophages and the formation of foam cells by regulating the ITCH/LKB1 axis by acting as a sponge for miR-17-5p, thus accelerating AS progression. The present study provides a theoretical basis for the development of novel therapeutic strategies for AS.
Methods
Collection of Clinical Samples
Carotid atherosclerotic plaques were collected from 10 AS patients (5 men, 5 women; mean [±SD] age 61.2±10.5 years) undergoing carotid endarterectomy operation in Chenzhou First People’s Hospital (20210816). The normal group consisted of 4 men and 6 women, with a mean (±SD) age of 58±8.5 years. Hypertensive patients without a significant history of liver or kidney disease were excluded from the control group. There were no statistically significant differences in age or sex between the AS and control groups. In addition, there were no statistically significant differences between the control and AS groups in terms of smoking history, diabetes, and chronic kidney disease, but there were significant difference in body mass index. Detailed characteristics are presented in the Supplementary Table.
Peripheral blood samples were collected from the normal group using vacuum blood collection tubes (anticoagulant tube/EDTA K2 · 2H2O). Blood samples were centrifuged with 1,006 g at 4℃ for 30 min, and monocytes were isolated from the blood for further analysis.
This study protocol was reviewed and approved by the Ethics Committee of Chenzhou First People’s Hospital, and all participants provided written informed consent.
Cell Culture and Treatment
Human monocytes (THP-1 cells), mouse mononuclear macrophages (RAW264.7 cells) and 293T cells, obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco) under 5% CO2
at 37℃. THP-1 cells were incubated with 100 nmol/L phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, St Louis, MO, USA) for 48 h at 37℃ to induce the differentiation of monocytes into macrophages (Mφ). When Mφ and RAW264.7 cells reached 70–80% confluence, they were stimulated in DMEM mixed with the fluorescently labeled Dil-oxLDL (10, 20, 30, 40 and 50 mg/L; Yeasen, Shanghai, China) for 48 h. For drug treatment, cells were treated with a concentration of cycloheximide (CHX; 5 μmol/L; MedChem Express, Monmouth Junction, NJ, USA) for 0, 1, 2, and 4 h or proteasome inhibitor MG132 (10 μmol/L; MedChem Express) for 6 h with 40 mg/L Dil-oxLDL for 48 h.
As reported in the Supplementary Methods, cells were subsequently used in transfection experiments examining the effects of NEAT1 and ITCH knockdown, as well as ITCH overexpression; coimmunoprecipitation (Co-IP) and RNA immunoprecipitation (RIP) assays; for the analysis of LKB1 ubiquitination; and lipid accumulation using Oil red O and BODIPY staining.
Animal Experiments
The ability of ApoE−/−
mice to clear lipoproteins is impaired and they easily form extensive fibrous plaque lesions in blood vessels. For the present study, 24 male ApoE−/−
mice (8 weeks old) were obtained from SJA LABORATORY Animal Co, Ltd. (Hunan, China). After acclimation for 1 week, mice were randomly divided into 3 groups: a control group, a short hairpin (sh)-negative control (NC) group, and a short hairpin (sh)-NEAT1 group (n=8 in each). ApoE−/−
mice were fed a high-fat diet (HFD; 21% fat + 0.15% cholesterol). The sh-NEAT1 adenovirus vector (Ad-sh-NEAT1) and Ad-sh-NC were constructed by GenePharma (Shanghai, China). After starting HFD feeding, mice in the sh-NC and sh-NEAT1 groups were injected with Ad-sh-NC and Ad-sh-NEAT1 (2×109
transduction units/mL) at 10 and 12 weeks, respectively, into the tail vein. After feeding of the HFD for 14 weeks, all mice were killed, and aortic tissue and blood samples were collected.
All experimental procedures were approved by the Ethics Committee of Chenzhou First People’s Hospital (20210813).
Statistical Analysis
All data from 3 independent experiments were used. Data are presented as the mean±SD and were analyzed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA). Student’s t-test or one-way analysis of variance (ANOVA) was used to determine the significance of differences among groups. Two-tailed P<0.05 was considered significant.
Results
NEAT1 and ITCH Were Up-Regulated, While miR-17-5p and LKB1 Were Down-Regulated
Carotid atherosclerotic plaques were collected from 10 AS patients. Expression of lncRNA NEAT1 and ITCH was significantly increased in the atherosclerotic plaques of AS patients compared with expression in control human macrophages, whereas the expression of miR-17-5p and LKB1 was reduced in the atherosclerotic plaques (Figure 1). These results suggest that NEAT1, miR-17-5p, ITCH and LKB1 are involved in AS progression.
Expression of NEAT1, ITCH, miR-17-5p, and LKB1 in the AS Cell Model
Foam cell formation, formed by the phagocytosis by macrophages in the early atherosclerotic plaques, is an important factor in AS progression.22 Macrophages formed by PMA-induced THP-1 cells (Mφ) and RAW264.7 cells were treated with different concentrations of Dil-oxLDL for 48 h. Oil red O staining showed that Dil-oxLDL increased lipid accumulation in macrophages in a concentration-dependent manner (Figure 2A). Dil-oxLDL 40 mg/L was selected for (40 mg/L) use in subsequent experiments. In addition, Dil-oxLDL treatment markedly upregulated lncRNA NEAT1 and ITCH in macrophages, but downregulated demonstrate miR-17-5p and LKB1 (Figure 2B,C). Collectively, the results that NEAT1, miR-17-5p, ITCH, and LKB1 were involved in foam cell formation during AS development.
Effects of ITCH Knockdown on Macrophage LDL Uptake and Lipid Accumulation
To investigate the role of ITCH in regulating foam cell formation during AS development, ITCH knockdown was induced in ox-LDL-treated macrophages. ITCH expression in macrophages was significantly reduced by short interfering (si)-ITCH transfection, indicating that the transfection was successful (Figure 3A). As shown in Figure 3B, Dil-oxLDL treatment significantly increased ITCH expression in macrophages, which was reversed by si-ITCH transfection. In addition, Dil-oxLDL stimulation enhanced LDL uptake by macrophages, which was reversed by ITCH knockdown (Figure 3C). Oil red O and BODIPY staining subsequently revealed that Dil-oxLDL stimulation increased lipid accumulation in macrophages, with this effect being abolished by ITCH silencing (Figure 3D,E). Together, the results indicate that the Dil-oxLDL-induced increases in LDL uptake by and lipid accumulation in macrophages were ameliorated by ITCH knockdown.

Role of LKB1 in the Effects of ITCH on Macrophage LDL Uptake and Lipid Accumulation
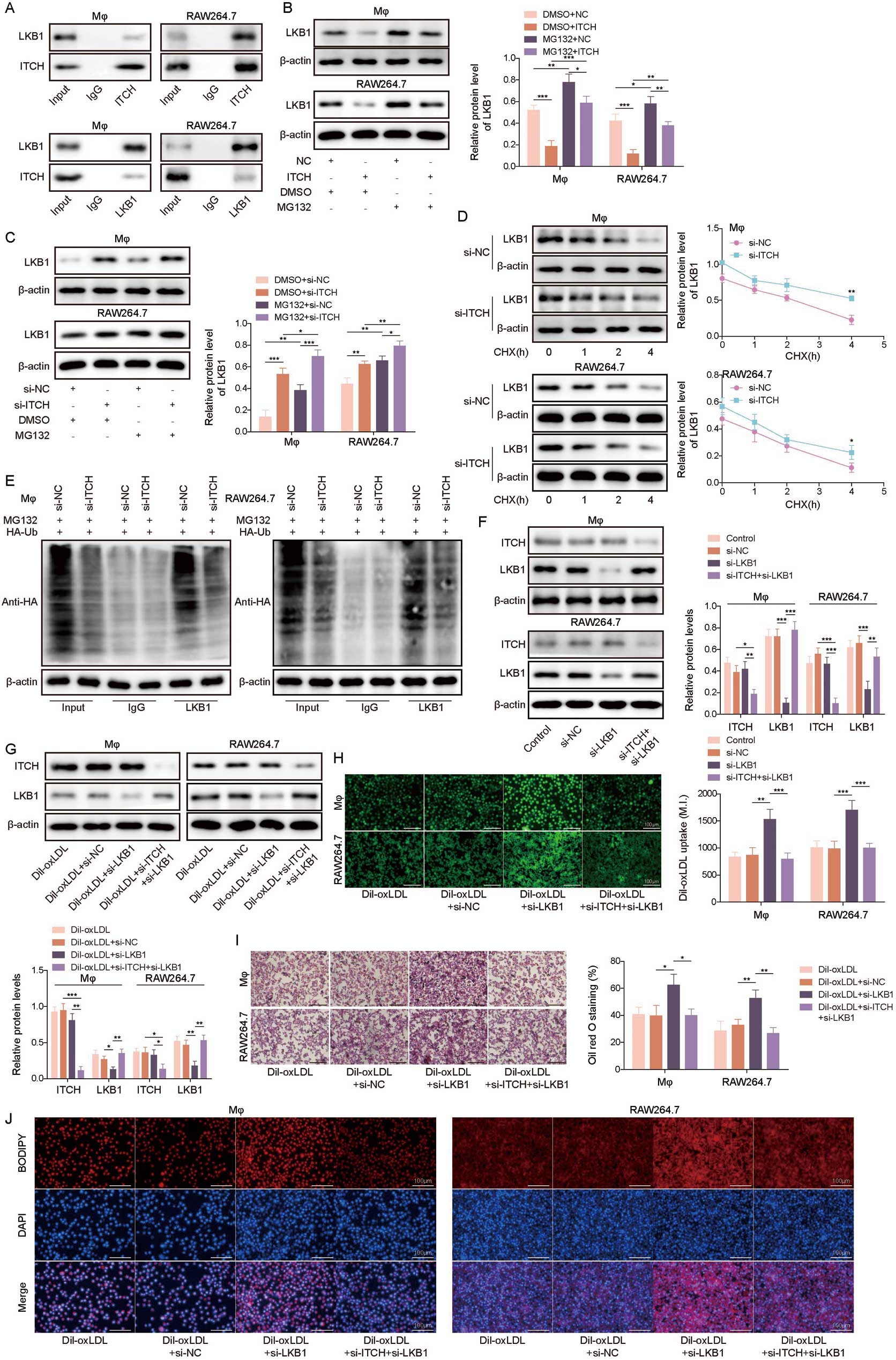
ITCH is an E3 ubiquitination ligase can mediate the ubiquitination of downstream targets and then regulate immune responses.23 In the present study, by using Co-IP assay, we found that ITCH directly bound to LKB1 (Figure 4A). In addition, ITCH overexpression reduced LKB1 levels in macrophages, with this effect being abrogated by treatment with MG132, a proteasome inhibitor (Figure 4B). ITCH knockdown increased LKB1 levels in macrophages, and the increase in LKB1 was more obvious in the presence of MG132 (Figure 4C). The CHX chase assay showed that LKB1 degradation in macrophages was slowed by ITCH knockdown (Figure 4D). Furthermore, LKB1 ubiquitination degradation in macrophages was reduced after ITCH silencing (Figure 4E). The transfection of Mφ and RAW264.7 cells transfected with si-LKB1 reduced LKB1 protein levels, while this change was reversed following si-ITCH transfection (Figure 4F). LKB1 protein levels in Dil-oxLDL-treated macrophages were reduced by si-LKB1 transfection, and this was reversed by ITCH silencing (Figure 4G). Functional experiments subsequently showed that LKB1 knockdown aggravated the Dil-oxLDL-induced increase in the uptake of LDL by and lipid accumulation in macrophages, whereas ITCH downregulation reversed the changes caused by LKB1 knockdown (Figure 4H–J). In summary, ITCH promoted the uptake of LDL by and lipid accumulation in macrophages during AS progression by facilitating ubiquitination degradation of LKB1.
Effects of NEAT1 Knockdown on Macrophage LDL Uptake and Lipid Accumulation
To investigate the role of the lncRNA NEAT1 in regulating foam cell formation during AS development, NEAT1 knockdown was induced in ox-LDL (40 mg/L)-treated macrophages by transfecting si-NEAT1 into cells. The transfection efficiency is shown in Figure 5A, and RT-qPCR results showed that si-NEAT1 transfection significantly reduced NEAT1 expression in Mφ and RAW264.7 cells. Dil-oxLDL treatment significantly increased NEAT1 expression in macrophages, and this change was reversed by si-NEAT1 transfection (Figure 5B). Functional experiments subsequently demonstrated that the uptake of LDL by and lipid accumulation in macrophages were enhanced by Dil-oxLDL stimulation, whereas these changes were abrogated by NEAT1 silencing (Figure 5C–E). Together, these results suggest that the Dil-oxLDL-induced increases in the uptake of LDL by and lipid accumulation in macrophages were ameliorated by NEAT1 knockdown.
Interactions Between NEAT1 and miR-17-5p, and Between miR-17-5p and ITCH
Interactions between NEAT1, miR-17-5p, and ITCH were subsequently investigated. Using starBase prediction, it was found that NEAT1 had potential binding sites for miR-17-5p (Figure 6A). The results of dual-luciferase reporter and RIP assays subsequently revealed that NEAT1 directly bound with miR-17-5p (Figure 6B,C). Meanwhile, it was predicted that miR-17-5p had potential binding sites for the 3′ untranslated region (UTR) of ITCH (Figure 6D). As confirmed by the dual-luciferase reporter and RIP assays, miR-17-5p directly bound with the 3′ UTR of ITCH (Figure 6E,F). In conclusion, NEAT1 directly bound with miR-17-5p, and miR-17-5p directly bound with ITCH.
Role of the miR-17-5p/ITCH/LKB1 Axis in the Effects of NEAT1 on Macrophage LDL Uptake and Lipid Accumulation
To the study the interaction between NEAT1 and the miR-17-5p/ITCH/LKB1 axis in regulating foam cell formation during AS progression, both miR-17-5p inhibition and NEAT1/ITCH knockdown were induced in Dil-oxLDL-treated macrophages. Suppression of miR-17-5p increased ITCH expression and reduced LKB1 expression in macrophages and Dil-oxLDL-treated macrophages, whereas NEAT1/ITCH knockdown alleviated the changes in ITCH and LKB1 expression caused by the miR-17-5p inhibitor (Figure 7A–D). In addition, miR-17-5p inhibition increased the uptake of LDL by and lipid accumulation in macrophages in the presence of Dil-oxLDL, whereas these changes were alleviated by NEAT1/ITCH knockdown (Figure 7E–G). In summary, NEAT1 facilitated the uptake of LDL by and lipid accumulation in macrophages during AS development by acting on the miR-17-5p/ITCH/LKB1 axis.
Effects of NEAT1 Knockdown on AS Progression In Vivo
To investigate the effects of NEAT1 on AS progression in vivo, we established AS model mice and knocked down NEAT1 in mice. NEAT1 knockdown reduced NEAT1 and ITCH expression and increased miR-17-5p and LKB1 expression in the aorta of AS model mice (Figure 8A–C). Serum total cholesterol and triglyceride concentrations were also analyzed, with NEAT1 knockdown having no significant effect on serum total cholesterol and triglyceride concentrations in AS model mice (Figure 8D). The results of histopathological analysis revealed that NEAT1 knockdown markedly ameliorated pathological changes and lipid accumulation in the aortic root lesions of AS model mice (Figure 8E,F). Collectively, these data prove that NEAT1 knockdown was effective in preventing AS progression in vivo.
Discussion
AS is a common cardiovascular disease and a high-risk factor for CAD and cerebral infarction, and effective treatment strategies for AS is lacking.24 It has been widely reported that AS is driven by the accumulation of cholesterol-rich macrophages (“foam cells”) in arterial walls,25 which is the main cause of CAD and cerebral infarction.26 Inhibiting foam cell formation may be a potential therapeutic strategy for AS. The primary novel findings in the present study are that upregulation of the lncRNA NEAT1 accelerates foam cell formation during AS development by regulating the miR-17-5p/ITCH/LKB1 axis.
Ubiquitination, as an important post-translational modification, has been widely involved in the pathogenesis of AS.27 Ubiquitination modification is regulated by ubiquitin-conjugating enzymes, ubiquitin ligases, and deubiquitinating enzymes. ITCH, as a ubiquitin ligase, play key roles in the pathogenesis of inflammatory diseases, including AS.28,29 As reported previously, ITCH deficiency could transform macrophages from a pro- to an anti-inflammatory phenotype and reduce HFD-induced lipid accumulation in macrophages.30 In addition, ITCH silencing reduced the formation of atherosclerotic plaques and circulating cholesterol concentrations in ApoE−/−
mice.20 In the present study, ITCH was significantly upregulated in the atherosclerotic plaques of AS patients and ox-LDL-treated macrophages, and its knockdown inhibited the uptake of LDL by and lipid accumulation in macrophages. The E3 ubiquitin ligase achieves its role in diseases by mediating the ubiquitination degradation of downstream targets. In the present study, ITCH mediated the ubiquitination degradation of LKB1 in macrophages by directly binding to LKB1. LKB1 has been shown to regulate macrophage function by mediating cell metabolism, growth, and polarization.31 Notably, recent studies have revealed the role of LKB1 in mediating the functional plasticity of macrophages in AS. For example, it was previously shown that LKB1 expression was reduced primarily in macrophages during AS progression, and its silencing exacerbated foam cell formation and AS progression by reducing scavenger receptor A phosphorylation and lysosome degradation.21 In addition, LKB1 downregulation in AS aggravated vascular damage by inducing excessive lipid deposition in macrophages and the formation of foam cells.31 Consistent with previous studies, our study showed that LKB1 expression was significantly decreased in the atherosclerotic plaques of AS patients and in ox-LDL-treated macrophages. In addition, as expected, LKB1 knockdown aggravated ox-LDL-induced increases in the uptake of LDL by and lipid accumulation in macrophages, whereas ITCH downregulation could reverse the changes caused by LKB1 knockdown. Collectively, our result show that ITCH upregulation accelerates foam cell formation and AS development by mediating the ubiquitination degradation of LKB1.
We subsequently investigated the upstream regulatory mechanism of the ITCH/LKB1 axis in regulating AS progression. Recent studies have revealed that lncRNAs are key players in AS progression.32 As proof, Li et al demonstrated that the expression of the lncRNA cyclin-dependent kinase inhibitor 2B antisense RNA 1 (CDKN2B-AS1) was reduced in atherosclerotic plaque tissue and THP-1-derived foam cells, and its overexpression inhibited lipid accumulation and cholesterol efflux in macrophages.33 In addition, lncRNA small nucleolar RNA host gene 16 (SNHG16) upregulation aggravated ox-LDL-induced macrophage proliferation and inflammatory response.16 The lncRNA NEAT1 has recently become a research hotspot in AS. As reported by Chen et al, NEAT1 expression in macrophages was increased by ox-LDL in a dose- and time-dependent manner, and its knockdown reduced ox-LDL-induced oxidative stress and inflammation in macrophages.34 It has also been reported that NEAT1 knockdown inhibited ox-LDL-induced inflammation and lipid uptake in macrophages.35 In the present study, NEAT1 was significantly upregulated in the atherosclerotic plaques of AS patients and ox-LDL-treated macrophages, which is consistent with the previous studies. Notably, NEAT1 knockdown reduced the uptake of LDL by and lipid accumulation in macrophages, as well as lesions and lipid accumulation in arterial tissues of AS mice. Our results suggest that NEAT1 facilitates lipid accumulation and foam cell formation in AS.
The target-mimetic, sponge/decoy function of lncRNAs on miRNAs in cancers has been widely reported.36 Nevertheless, it is unknown whether NEAT1 plays its regulatory role in AS by acting as a sponge of miRNA. Herein, we proposed a new ceRNA regulatory network in AS, in which the lncRNA NEAT1 acted as a sponge for miR-17-5p. miR-17-5p is a tumor suppressor gene that exerts anti-angiogenic activity.37 According to reports, miR-17-5p play dual roles in AS. First, miR-17-5p inhibition reduces inflammation and lipid accumulation in macrophages during AS development.38 However, another study showed that miR-17-5p overexpression inhibits ox-LDL-induced cell proliferation and inflammatory responses in macrophages.16 In the present study, miR-17-5p was downregulated in the atherosclerotic plaques of AS patients and ox-LDL-treated macrophages. We subsequently revealed that NEAT1 functioned as a sponge for miR-17-5p to negatively regulate miR-17-5p expression in macrophages, and that miR-17-5p negatively regulated ITCH in macrophages by directly targeting ITCH. As expected, miR-17-5p inhibition increased the uptake of LDL by and lipid accumulation in macrophages in the presence of Dil-oxLDL, whereas these changes were alleviated by NEAT1/ITCH knockdown. Therefore, we came to the conclusion that NEAT1 regulated the ITCH/LKB1 axis by acting as a sink/sponge for miR-17-5p to promote foam cell formation and AS progression.
Together, our study, for the first time, demonstrate that the lncRNA NEAT1 accelerates foam cell formation during AS progression through the miR-17-5p/ITCH/LKB1 axis. Our study provides a theoretical basis for the development of novel therapeutic strategies for AS.
Acknowledgments
Not applicable.
Sources of Funding
This work was supported by the Research Plan Project of Hunan Provincial Health Commission (202202052843).
Disclosures
The authors declare that there is no conflict of interest.
Author Contributions
H.H. designed the experiments and wrote the manuscript. B.P. performed most of the experiments. Q.C. performed some of the experiments. Y.W. analyzed the data and organized the figures. R.L. revised the article.
IRB Information
The study protocol was reviewed and approved by the Ethics Committee of Chenzhou First People’s Hospital. All participants provided written informed consent. All animal experimental procedures were approved by the Ethics Committee of Chenzhou First People’s Hospital (20210813).
Data Availability
The datasets generated and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Supplementary Files
Please find supplementary file(s);
https://doi.org/10.1253/circj.CJ-23-0769
References
- 1.
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics – 2022 update: A report from the American Heart Association. Circulation 2022; 145: e153–e639.
- 2.
Poznyak AV, Bharadwaj D, Prasad G, Grechko AV, Sazonova MA, Orekhov AN. Renin-angiotensin system in pathogenesis of atherosclerosis and treatment of CVD. Int J Mol Sci 2021; 22: 6702, doi:10.3390/ijms22136702.
- 3.
Bobryshev YV, Ivanova EA, Chistiakov DA, Nikiforov NG, Orekhov AN. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. Biomed Res Int 2016; 2016: 9582430.
- 4.
Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol 2020; 250: 705–714.
- 5.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet 2006; 15(Spec No 1): R17–R29.
- 6.
Zhang P, Wu S, He Y, Li X, Zhu Y, Lin X, et al. LncRNA-mediated adipogenesis in different adipocytes. Int J Mol Sci 2022; 23: 7488, doi:10.3390/ijms23137488.
- 7.
Fasolo F, Di Gregoli K, Maegdefessel L, Johnson JL. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc Res 2019; 115: 1732–1756.
- 8.
Tang X, Yin R, Shi H, Wang X, Shen D, Wang X, et al. LncRNA ZFAS1 confers inflammatory responses and reduces cholesterol efflux in atherosclerosis through regulating miR-654-3p-ADAM10/RAB22A axis. Int J Cardiol 2020; 315: 72–80.
- 9.
Zhang X, Guan MX, Jiang QH, Li S, Zhang HY, Wu ZG, et al. NEAT1 knockdown suppresses endothelial cell proliferation and induces apoptosis by regulating miR-638/AKT/mTOR signaling in atherosclerosis. Oncol Rep 2020; 44: 115–125.
- 10.
Wang L, Xia JW, Ke ZP, Zhang BH. Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR-342-3p in human macrophages THP-1 cells. J Cell Physiol 2019; 234: 5319–5326.
- 11.
Hill M, Tran N. miRNA interplay: Mechanisms and consequences in cancer. Dis Model Mech 2021; 14: dmm047662, doi:10.1242/dmm.047662.
- 12.
Lu Y, Thavarajah T, Gu W, Cai J, Xu Q. Impact of miRNA in atherosclerosis. Arterioscler Thromb Vasc Biol 2018; 38: e159–e170.
- 13.
Wang J, Zhang L, Wang T, Li C, Jiao L, Zhao Z, et al. miRNA-576 alleviates the malignant progression of atherosclerosis through downregulating KLF5. Dis Markers 2021; 2021: 5450685.
- 14.
Zhang H, Hao J, Sun X, Zhang Y, Wei Q. Circulating pro-angiogenic micro-ribonucleic acid in patients with coronary heart disease. Interact Cardiovasc Thorac Surg 2018; 27: 336–342.
- 15.
Chen J, Xu L, Hu Q, Yang S, Zhang B, Jiang H. MiR-17-5p as circulating biomarkers for the severity of coronary atherosclerosis in coronary artery disease. Int J Cardiol 2015; 197: 123–124.
- 16.
An JH, Chen ZY, Ma QL, Wang HJ, Zhang JQ, Shi FW. LncRNA SNHG16 promoted proliferation and inflammatory response of macrophages through miR-17-5p/NF-κB signaling pathway in patients with atherosclerosis. Eur Rev Med Pharmacol Sci 2019; 23: 8665–8677.
- 17.
Ma N, Tie C, Yu B, Zhang W, Wan J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging (Albany NY) 2020; 12: 2897–2920.
- 18.
Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J 2005; 24: 836–848.
- 19.
Poels K, Vos WG, Lutgens E, Seijkens TTP. E3 ubiquitin ligases as immunotherapeutic target in atherosclerotic cardiovascular disease. Front Cardiovasc Med 2020; 7: 106.
- 20.
Stöhr R, Mavilio M, Marino A, Casagrande V, Kappel B, Möllmann J, et al. ITCH modulates SIRT6 and SREBP2 to influence lipid metabolism and atherosclerosis in ApoE null mice. Sci Rep 2015; 5: 9023.
- 21.
Liu Z, Zhu H, Dai X, Wang C, Ding Y, Song P, et al. Macrophage liver kinase B1 inhibits foam cell formation and atherosclerosis. Circ Res 2017; 121: 1047–1057.
- 22.
Johnston JM, Angyal A, Bauer RC, Hamby S, Suvarna SK, Baidžajevas K, et al. Myeloid Tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci Adv 2019; 5: eaax9183.
- 23.
Field NS, Moser EK, Oliver PM. Itch regulation of innate and adaptive immune responses in mice and humans. J Leukoc Biol 2020; 108: 353–362.
- 24.
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011; 473: 317–325.
- 25.
Zhang X, McDonald JG, Aryal B, Canfrán-Duque A, Goldberg EL, Araldi E, et al. Desmosterol suppresses macrophage inflammasome activation and protects against vascular inflammation and atherosclerosis. Proc Natl Acad Sci USA 2021; 118: e2107682118, doi:10.1073/pnas.2107682118.
- 26.
Kotla S, Singh NK, Rao GN. ROS via BTK-p300-STAT1-PPARγ signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol 2017; 11: 350–364.
- 27.
Zhou ZX, Ren Z, Yan BJ, Qu SL, Tang ZH, Wei DH, et al. The role of ubiquitin E3 ligase in atherosclerosis. Curr Med Chem 2021; 28: 152–168.
- 28.
Lu Y, Zhang X, Hu W, Yang Q. The identification of candidate biomarkers and pathways in atherosclerosis by integrated bioinformatics analysis. Comput Math Methods Med 2021; 2021: 6276480.
- 29.
Theivanthiran B, Kathania M, Zeng M, Anguiano E, Basrur V, Vandergriff T, et al. The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci Signal 2015; 8: ra22.
- 30.
Marino A, Menghini R, Fabrizi M, Casagrande V, Mavilio M, Stoehr R, et al. ITCH deficiency protects from diet-induced obesity. Diabetes 2014; 63: 550–561.
- 31.
Wang X, Liang Z, Xiang H, Li Y, Chen S, Lu H. LKB1 regulates vascular macrophage functions in atherosclerosis. Front Pharmacol 2021; 12: 810224.
- 32.
Abd-Elmawla MA, Fawzy MW, Rizk SM, Shaheen AA. Role of long non-coding RNAs expression (ANRIL, NOS3-AS, and APOA1-AS) in development of atherosclerosis in Egyptian systemic lupus erythematosus patients. Clin Rheumatol 2018; 37: 3319–3328.
- 33.
Li H, Han S, Sun Q, Yao Y, Li S, Yuan C, et al. Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany NY) 2019; 11: 1695–1715.
- 34.
Chen DD, Hui LL, Zhang XC, Chang Q. NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128. J Cell Biochem 2019; 120: 2493–2501.
- 35.
Huang-Fu N, Cheng JS, Wang Y, Li ZW, Wang SH. Neat1 regulates oxidized low-density lipoprotein-induced inflammation and lipid uptake in macrophages via paraspeckle formation. Mol Med Rep 2018; 17: 3092–3098.
- 36.
Paraskevopoulou MD, Hatzigeorgiou AG. Analyzing miRNA-lncRNA interactions. Methods Mol Biol 2016; 1402: 271–286.
- 37.
Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 2006; 38: 1060–1065.
- 38.
Tan L, Liu L, Jiang Z, Hao X. Inhibition of microRNA-17-5p reduces the inflammation and lipid accumulation, and up-regulates ATP-binding cassette transporterA1 in atherosclerosis. J Pharmacol Sci 2019; 139: 280–288.






