Abstract
Background: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease with a poor prognosis and no curative therapy. It may present as arrhythmogenic sudden cardiac death and inevitably progress to terminal heart failure due to the loss of contractile tissue. This study aimed to generate knock-in (KI) mice carrying the 2 genetic variants (DSG2 p.R292C and p.D494A) most frequently found in Japanese ARVC patients, characterize their cardiac phenotype, and compare the results with those of human ARVC.
Methods and Results: Variants were introduced using CRISPR/Cas9 genome editing at the corresponding mouse locations: Dsg2 p.R297C (RC) and p.D499A (DA). Cardiac function, morphology, and electrophysiology were evaluated using echography, magnetic resonance imaging, and telemetry. Tissue and cardiomyocytes were examined histologically. All mice with the variants developed biventricular cardiac dysfunction after 8 weeks of age, and it progressed with age. There was a significant variability in phenotype expression. Mice with RC died suddenly at 9 weeks of age. Some homozygous RC mice showed arrhythmia and conduction abnormalities on telemetry. In both variants, staining of cardiac sections revealed significant fibrosis, and apoptosis was detected using the terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling assay.
Conclusions: We generated a KI ARVC mouse model with significant similarities to human disease. This model could be used for the elucidation of pathogenesis and the development of optimal therapy for ARVC.

Central Figure
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disease with a population-dependent prevalence of 1 : 1,000–1 : 5,000. ARVC causes structural cardiac damage predominantly in the right ventricle, electrical instability, and heart failure.1 Cardiomyocyte death and fibrofatty replacement of the myocardium are the hallmark findings in cardiac biopsies and explanted ARVC hearts.2 Malignant ventricular arrhythmias, specifically during strenuous physical exercise, and particularly in athletes, and sudden cardiac death (SCD) can be the initial clinical presentation of ARVC. During disease progression, the left ventricle is also affected, and finally terminal heart failure develops. The diagnosis of ARVC is based on the Task Force Criteria.3 The treatment of ARVC targets only clinical manifestations, and curative therapy has not yet been discovered.1 Therefore, ARVC has been actively investigated, both clinically and translationally.
In ARVC, the desmosome is the key factor: plakophillin-2, desmoglein-2, desmoplakin, desmocollin-2, and junctional plakoglobin are the 5 core components of desmosome organelles, providing their main functionality in maintaining the structural integrity of the heart muscle.1 These desmosomal proteins are encoded by PKP2, DSG2, DSP, DSC2, and JUP. Nearly 60% of ARVC patients carry pathogenic variants, and mostly in these desmosomal genes. Various biological processes depend on proper desmosome assembly. Heritable ARVC variants are typically autosomal dominant and exhibit incomplete penetrance. In Caucasian ARVC patients, the most prevalent heterozygous variants are of PKP2, followed by DSG2.4 In East Asian countries, DSG2 variants are the most frequently found, especially in the compound heterozygous or homozygous mode: DSG2 p.D292C and p.D494A are the most common in Japan, and p.F531C in China.5,6 In an early report of desmosomal variants with compound heterozygous or homozygous patterns, the skin and hair of patients with these patterns were affected, in addition to cardiac phenotypes such as Naxos disease and Carvajal syndrome.7,8 In contrast, ARVC patients with biallelic DSG2 variants in East Asia rarely exhibit skin and hair abnormalities.5,6
We focused on the investigation of Dsg2 knock-in (KI) mouse models because of the indispensable significance of desmoglein-2 for mechanical strength of cardiac tissue, and the fact that DSG2 variants are the most prominent among Japanese ARVC patients.
This study aimed to generate Dsg2 KI mouse models that harbor the most common genetic variants in Japanese ARVC patients, p.R292C and p.D494A. We selected the 2 most frequent missense variants in the Japanese ARVC cohort corresponding to p.R297C (RC) and p.D499A (DA) in mice and compared the mouse phenotypes with the known characteristics of ARVC patients.
Methods
A detailed description of the methods is provided in the Supplementary Information. The timeline of the experiments is summarized in Supplementary Figure 1.
Generation of Dsg2 KI Mice and Validation of the Variants by Sanger Sequencing
All animal experiments were conducted according to the ethical requirements of the National and Cerebrovascular Centre and were approved by protocol no. 20025. KI mouse models with missense variants of Dsg2 (Supplementary Figure 2A) based on the B6D2F1 strain were generated using CRISPR/Cas9 genome editing following a previously reported protocol.9 To verify base substitution, genomic DNA was isolated and analyzed by Sanger sequencing using SeqStudio (Applied Biosystems, Thermo Fisher Scientific, MA, USA) (Supplementary Figure 2B).
Histological Analysis
The hearts of wild-type (WT), RC, and DA mice, half-cut through the short axis of the ventricles, were embedded in paraffin blocks. Cardiac sections, 4-µm thick, were made using a Leica RM2125RT microtome (Leica Microsystems, Germany) and attached to a micro-slide glass (Matsunami, Japan) coated with poly-L-lysine. Standard protocols for hematoxylin and eosin (HE) and Masson’s trichrome staining were used to stain the paraffin sections.
Statistical Analysis
The statistical functions of IgorPro 9.0 (WaveMetrics Inc., OR, USA), GraphPad Prism 5 (GraphPad software, MA, USA) and R version 4.3.3 were applied to assess the summarized experimental results. Numerical data are shown as mean±standard deviation. Differences between 2 groups were examined using Student’s t-test. Three groups of data were evaluated using one-way analysis of variance, followed by Bonferroni’s post hoc test. Statistical significance was set at P<0.05.
Results
Growth, Life Expectancy, and Macroscopic Findings of Dsg2 KI Mice
KI mice grew indistinguishably from WT mice. However, the heart/body weight ratio of homozygous RC mice at 45 weeks was significantly higher than that of WT mice (Supplementary Table 1). We observed SCD in mice with homozygous RC starting at the age of 9 weeks (Supplementary Figure 3A). The Kaplan-Meier estimator for the duration of 50 weeks did not show death of WT, homozygous, or heterozygous DA mice; in contrast, the mean survival of heterozygous and homozygous RC mice was ~80% and ~60%, respectively, and the P value of the log-rank test was 0.032 (Supplementary Figure 3A). Supplementary Figure 3B shows the macroscopic hearts of WT mice; RC mice died suddenly at 12 and 9 weeks of age. Compared with the hearts of WT mice, those of the suddenly dead RC mice were enlarged, predominantly due to the expanded right atria (blue arrow) and ventricles (green arrows). In most mice with SCD, the myocardium had diffusely scattered pale areas suggestive of a damaged region (Supplementary Figure 3B, arrowheads).
Collagen Accumulation and Calcification in the Myocardial Tissue of Dsg2 Variants
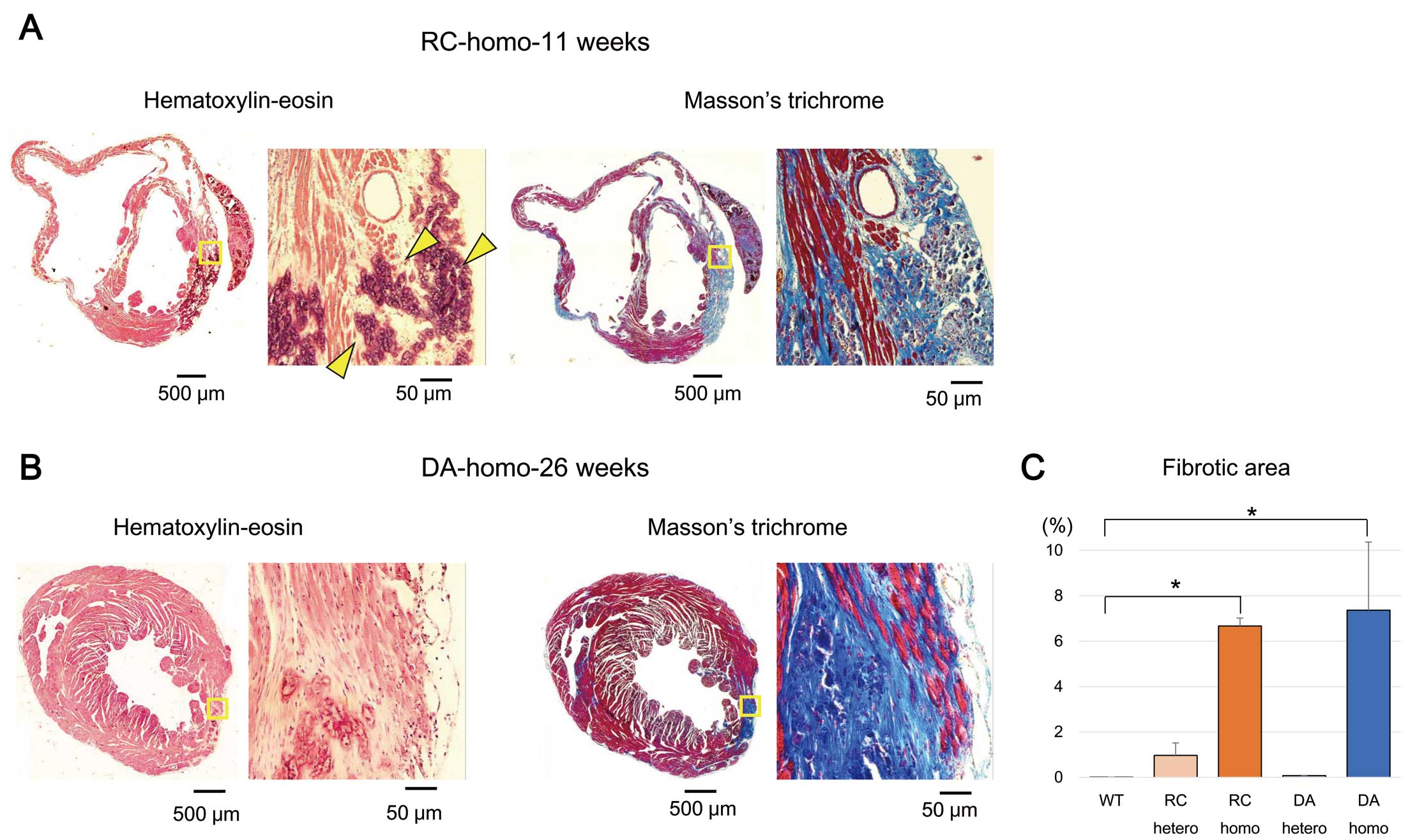
Visible morphological lesions in the myocardium of mice that died suddenly, as early as 9 weeks of age (Supplementary Figure 3B), were also observed in the hearts of killed mice at various ages. The hearts of killed WT and KI mice were embedded in paraffin blocks, and cardiac sections were stained with HE, Masson’s trichrome, and Gomori trichrome to reveal the gross morphology and collagen content of the myocardium. The last 2 stains were used to differentially stain collagen and muscle fibers. Heart sections from 26-week-old WT mice showed morphological properties similar to those of healthy hearts and were negative for collagen (Supplementary Figure 4A). The sections from mice with heterozygous RC or DA stained with HE had a morphology similar to that of WT mice, but a small amount of collagen developed around the vessels and subepicardial tissue, as revealed by Masson’s trichrome staining (Supplementary Figure 4B,C). In contrast, in HE-stained sections of 11-week-old RC homozygous mice (Figure 1A), microscopic violet-blue aggregations were clearly observed, representing the typical staining of calcium deposits by hematoxylin. Masson’s trichrome staining of the same section showed massive fibrosis in weakly HE-stained and calcium-containing areas. In a section from a 19-week-old killed mouse with homozygous RC, a segment of the left ventricular wall was completely replaced with fibrotic tissue (Supplementary Figure 5A). Gomori trichrome staining of a section from a 26-week-old RC homozygous mouse (Supplementary Figure 5B) revealed massive fibrosis and calcium deposits. Figure 1B shows HE and Masson’s trichrome staining of homozygous DA mice at 26 weeks of age. Broad fibrotic areas can be observed, though the progression of fibrosis was slower than for RC. Figure 1C summarizes the quantification of fibrotic areas in the cardiac sections of both homozygous and heterozygous mice at 25–30 weeks of age. The percentages of fibrotic areas in homozygous heart sections were significantly increased compared with those in the hearts of WT mice (6.7±0.35% and 7.4±3.0% for homozygous RC and DA, respectively). In heterozygous mice, the fibrotic area in the RC hearts was slightly increased (0.97±0.55%) and was almost the same in DA (0.07±0.01%) as WT. These fibrotic areas were prevalent in the mid to basal areas and rarely observed in the apex.
Cardiac Functional and Morphological Changes in Dsg2 KI Mice
Cardiac function and morphology were assessed using echocardiography and magnetic resonance imaging (MRI).
Echocardiography was performed in adult mice of various ages starting at 9 weeks old (Supplementary Table 2). Decline in left and right ventricular function was observed in all KI mice, with significant variability that worsened with age (Supplementary Tables 2,3). WT mice maintained normal heart size and function in both ventricles (Figure 2A). The left ventricular ejection fraction (LVEF) and tricuspid annular plane systolic excursion (TAPSE) in RC heterozygous mice at 45 weeks of age showed a mild decrease (Figure 2B). Homozygous DA mice exhibited enlarged right ventricles and decreased cardiac function (Figure 2C, Supplementary Figure 6). Figure 2D,E summarizes the LVEF and TAPSE values of homozygous and heterozygous KI mice at 9 and 45 weeks, respectively. The echocardiographic parameters are presented in Supplementary Table 2 and Supplementary Table 3. Phenotypic variability is clearly visible in Supplementary Figure 6, showing a significant difference in LV function between 2 homozygous DA mice at the age of 90 weeks.

These results were confirmed by cardiac MRI measurements of chamber volumes and functions in 16–19-week-old RC mice. Compared with WT mice (Figure 3A, Left), homozygous (Figure 3A, Right) mice showed significantly decreased EFs in both ventricles, although the end-diastolic volumes of both ventricles were not significantly different. These data are summarized in Figure 3B and Supplementary Table 4.
In both echocardiography and cardiac MRI, we did not observe obvious aneurysm, dyskinesis or akinesis in any of the KI mice, though it was difficult to detect subtle changes in morphology. Overall, these 2 Dsg2 variants induced cardiac dysfunction and enlargement in our mouse model.
Arrhythmias Recorded by Telemetric ECG Recording
As shown in the disease name, ARVC is characterized by ventricular arrhythmias originating from the right ventricles. We evaluated the occurrence of arrhythmias through continuous ECG recordings using a telemetry system. A total of 4 WT, heterozygous, and homozygous RC mice per group were implanted with a subcutaneous telemetry transmitter. Evaluation of the recordings revealed intermittent electrical abnormalities in 2 homozygous RC mice. In 1 homozygous 15-week-old mouse, second-degree atrioventricular block was recorded (Supplementary Figure 7A). Premature ventricular contractions and tachycardia were observed in 15- and 18-week-old homozygous mice (Supplementary Figure 7B–D). In other mice, including 2 17-week-old homozygous mice, no abnormal electrical findings, including abnormal electrical conduction, were observed (Supplementary Figure 7E, Supplementary Table 5).
Cellular Characteristics of the Myocardium in KI Mice
To evaluate the apoptosis of cardiomyocytes affected by Dsg2 variants, we performed nuclear staining using terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL). In the positive control with WT mice, we observed clear red staining, indicating apoptosis (Figure 4A), but not in the negative control (Figure 4B). In samples from both heterozygous and homozygous KI mice, red staining suggested apoptotic processes related to Dsg2 variants (Figure 4C,D). Figure 4E shows the number of TUNEL-positive nuclei as follows: 0 positive from 126 WT nuclei, 42 positive from 353 RC heterozygous cells, 42 positive from 183 RC homozygous cells, 21 positive from 308 DA heterozygous cells, and 17 positive from 113 DA homozygous cells, suggesting a higher percentage of apoptosis in the samples from KI mice than in those from WT mice.
Discussion
We generated KI mouse models of 2 DSG2 variants, p.R292C and p.D494A, that are frequently found in Japanese ARVC patients. These models present the most typical DSG2 missense variants found in human ARVC.6 Similar to human ARVC caused by DSG2 variants,10 our Dsg2 KI mouse models showed biventricular dysfunction in the early phase of the disease. In contrast, aneurysm and dyskinesis of the ventricles, which have been included in the ARVC diagnostic criteria,3 were hardly detected in the KI mouse models. These morphological changes were supported by histological analysis of KI mouse models, which scarcely showed fibrotic changes in the apex. In addition, ventricular arrhythmias simultaneously observed in human ARVC caused by DSG2 variants11 were rarely observed.
Desmoglein-2 has several physiological functions. Inactivation of desmoglein-2 in embryonic stem cells produces defects in blastocysts, and prevents cell proliferation and embryonic survival in mice, demonstrating its indispensable role in embryonic viability.12 Numerous cellular and mouse models have been developed to demonstrate the effects of desmoglein-2 dysfunction. Using the HL-1 cardiomyocyte cell line, Schlipp et al. demonstrated that ARVC-related mutations in DSG2 disrupted the cohesion between cardiomyocytes, similar to L-tryptophan treatment.13 Murine ARVC models have been generated using different techniques to interfere with desmoglein-2 function. Pilichou et al. overexpressed Dsg2 p.N271S in the mouse heart, equivalent to DSG2 p.N266S, and found that mice developed cardiac necrosis as the basis for further myocardial dystrophy.14 Rizzo et al. used transgenic mice with cardiac overexpression of Dsg2 p.N271S to show electrical and structural abnormalities in the intercalated disks, which are critical membrane structures located on the short side of cardiomyocytes that maintain mechanical and electrical integrity in the heart.15Dsg2 p.N271S suppressed the peak sodium current, slowed conduction, and widened intercellular spaces in desmosomal areas. Other experiments have manipulated the desmoglein-2 architecture in the heart. Kant et al. showed a loss of adhesive function between cardiomyocytes and accumulation of skeletal actin in intercalated disks in both heart-specific Dsg2 knockdown and lacking extracellular domain mouse models.16 Zhang et al. constructed a Dsg2 KI mouse model with p.F536C, corresponding to DSG2 p.F531C highly identified Chinese ARVC, and found a significant effect of ATF4/TGF-β1 signaling for cardiac fibrosis.17
Although our models had only 1 amino acid substitution, we observed SCD (Supplementary Figure 3), development of fibrosis (Figure 1, Supplementary Figures 4,5), heart failure and cardiac enlargement (Figures 2,3), arrhythmia and conduction abnormalities (Supplementary Figure 7), and apoptosis (Figure 4) in RC mice, as in the aforementioned transgenic mice possessing significantly more damaged desmoglein-2. In particular, the functional and structural processes produced by Dsg2 variants were more severe in the hearts of homozygous mice.
Desmoglein-2 interacts in a trans position via the tryptophan swap, which is the second amino acid of the extracellular domain 1.18 The third and fourth extracellular domains of desmoglein-2, where the RC and DA variants are located, are not directly involved in homophilic or heterophilic desmoglein-2 interactions. In ARVC, the common understanding is that desmosome dysfunction weakens cell-cell connections, leading to cardiomyocyte damage and a characteristic tissue appearance. The Dsg2 variants investigated in this study may indirectly affect desmoglein-2 trans interactions or disturb the inherent flexibility of the extracellular domain of the molecule,18 thus causing detrimental cellular effects.
Our 2 KI mouse models similarly showed fibrotic changes in the myocardium (Figure 1) and RV dysfunction (Figure 2E). When we compared the 2 models, RC homozygous mice showed a more severe phenotype than DC homozygous mice (Supplementary Figure 3A). The fibrotic changes and cardiac functions in RC homozygous mice were those that survived until the age of evaluation, and severely affected RC homozygous mice were eliminated. The difference in severity between the 2 KI mice models might be related to the location of the variants. A previous study reported that variants in cadherin repeat domains 1–3 were highly identified in ARVC patients but not in domain 4.19 The domain of the cadherin repeat is different in the 2 variants: RC in domain 3 and DA in domain 4.19
In our ARVC registration database, we identified homozygous p.R292C in 12 patients and p.D494A in 13 patients. There was no significant difference in the ages of onset or cardiac function between these homozygous variant carriers.11 However, among 11 ARVC patients with DSG2 variants and aged <20 years old, 3 patients carried homozygous p.R292C, and no one carried homozygous p.D494A. The data from our ARVC database seems to correspond to our KI mouse data, whereby homozygous RC mice showed a younger onset than those of DA.
ARVC is characterized by ventricular arrhythmias originating from the right ventricle;1 however, ventricular arrhythmias were rarely observed in our mouse model (Supplementary Figure 7, Supplementary Table 5). In contrast, an ARVC mouse model with a Pkp2 variant showed frequent ventricular arrhythmias.20 These genotype-dependent phenotypic differences have been reported in human ARVC; DSG2 variant carriers have a higher risk of heart failure than PKP2 variant carriers.21
The treatments for ARVC at present are to prevent ventricular arrhythmias and to relieve the symptoms due to heart failure, and they are not a molecular-based treatment. In obstructive hypertrophic cardiomyopathy, mavacamten, which inhibits myosin binding, showed significant improvements in clinical evaluation.22 Although we have not found the therapeutic target for ARVC, our KI mouse models should be useful for the development of molecular-based treatment for ARVC.
The goal of this study was to generate mouse models harboring patient-specific variants of ARVC; the cardiac phenotype produced was similar to that of human ARVC. We observed significant similarities to human disease in our KI mice: SCD in RC mice; cardiac fibrosis in both variants, particularly in homozygous mice; chamber enlargement and heart failure that progressed with age; and lethal arrhythmia in only a small number of RC homozygous mice.
Study Limitations
Our ARVC mouse models resembled the human phenotype; however, we did not identify molecules that affect the degeneration of desmoglein-2 or the main mechanisms of ARVC that drive the mouse phenotype, which may differ from human phenotypes.
Conclusions
Our models produced a distinctive phenotype resembling that of human ARVC. This allows for the use of KI mice for the elucidation of the pathogenesis of ARVC. In addition, KI mice could be useful for the screening of pharmaceutical agents that may improve or even prevent pathophysiological and morphological alterations in KI mouse hearts. Successful experimental data can be used as a step towards the introduction of new drugs with curative capabilities for ARVC.
Acknowledgments
We thank Ms. Manami Sone for the excellent preparation of paraffin sections. This study was supported by a grant-in-aid for Scientific Research, Ministry of Education, Culture, Sports, Science, and Technology of Japan, KAKENHI for D.P.Z. and S.O. (21K08119).
Disclosures
The authors declare no competing interests relevant to the contents of this article.
IRB Information
Not applicable.
Author Contributions
S.O., D.P.Z.: conceptualization, funding acquisition, writing the original draft, formal analysis, writing-review and editing; D.P.Z., H.T., S.S., M.Z., Y.F.: investigation and resources; M.B., K.S.: validation and data curation. All authors have read and approved the final manuscript.
Supplementary Files
Please find supplementary file(s);
https://doi.org/10.1253/circj.CJ-25-0269
References
- 1.
Krahn AD, Wilde AAM, Calkins H, La Gerche A, Cadrin-Tourigny J, Roberts JD, et al. Arrhythmogenic right ventricular cardiomyopathy. JACC Clin Electrophysiol 2022; 8: 533–553.
- 2.
Asimaki A, Saffitz JE. The role of endomyocardial biopsy in ARVC: Looking beyond histology in search of new diagnostic markers. J Cardiovasc Electrophysiol 2011; 22: 111–117.
- 3.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur Heart J 2010; 31: 806–814.
- 4.
Imai Y, Kusano K, Aiba T, Ako J, Asano Y, Harada-Shiba M, et al. JCS/JCC/JSPCCS 2024 Guideline on genetic testing and counseling in cardiovascular disease. Circ J 2024; 88: 2022–2099.
- 5.
Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pilichou K, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2010; 55: 587–597.
- 6.
Wada Y, Ohno S, Aiba T, Horie M. Unique genetic background and outcome of non-Caucasian Japanese probands with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Mol Genet Genom Med 2017; 5: 639–651.
- 7.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000; 355: 2119–2124.
- 8.
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas JC, Common J, Purkis PE, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000; 9: 2761–2766.
- 9.
Fujihara Y, Ikawa M. CRISPR/Cas9-based genome editing in mice by single plasmid injection. Methods Enzymol 2014; 546: 319–336.
- 10.
Chen L, Hu Y, Saguner AM, Bauce B, Liu Y, Shi A, et al. Natural history and clinical outcomes of patients with DSG2/DSC2 variant-related arrhythmogenic right ventricular cardiomyopathy. Circulation 2025; 151: 1213–1230.
- 11.
Sonoda K, Nagase S, Aiba T, Kato K, Fukuyama M, Kikuchi N, et al. Homozygous or compound heterozygous variants in DSG2 are mainly causative of Japanese arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2023; 44: ehad655.612.
- 12.
Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol 2002; 81: 592–598.
- 13.
Schlipp A, Schinner C, Spindler V, Vielmuth F, Gehmlich K, Syrris P, et al. Desmoglein-2 interaction is crucial for cardiomyocyte cohesion and function. Cardiovasc Res 2014; 104: 245–257.
- 14.
Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, et al. Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med 2009; 206: 1787–1802.
- 15.
Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, et al. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res 2012; 95: 409–418.
- 16.
Kant S, Holthofer B, Magin TM, Krusche CA, Leube RE. Desmoglein 2-dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ Cardiovasc Genet 2015; 8: 553–563.
- 17.
Zhang B, Wu Y, Zhou C, Xie J, Zhang Y, Yang X, et al. Hyperactivation of ATF4/TGF-β1 signaling contributes to the progressive cardiac fibrosis in arrhythmogenic cardiomyopathy caused by DSG2 Variant. BMC Med 2024; 22: 361.
- 18.
Tariq H, Bella J, Jowitt TA, Holmes DF, Rouhi M, Nie Z, et al. Cadherin flexibility provides a key difference between desmosomes and adherens junctions. Proc Natl Acad Sci U S A 2015; 112: 5395–5400.
- 19.
Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol 2011; 57: 2317–2327.
- 20.
Bradford WH, Zhang J, Gutierrez-Lara EJ, Liang Y, Do A, Wang TM, et al. Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics. Nat Cardiovasc Res 2023; 2: 1246–1261.
- 21.
Hermida A, Fressart V, Hidden-Lucet F, Donal E, Probst V, Deharo JC, et al. High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Heart Fail 2019; 21: 792–800.
- 22.
Kitaoka H, Ieda M, Ebato M, Kozuma K, Takayama M, Tanno K, et al. Phase 3 open-label study evaluating the efficacy and safety of mavacamten in Japanese adults with obstructive hypertrophic cardiomyopathy: The HORIZON-HCM study. Circ J 2025; 89: 130–138.

