症例報告
IgG4関連疾患に見られた再発性脊髄炎の1例
2021 年 61 巻 12 号 p. 839-843

詳細
2021 年 61 巻 12 号 p. 839-843
患者は80歳男性.IgG4関連呼吸器疾患で2年間のステロイド維持治療中に,腹部以下の痺れ,感覚失調性の歩行障害が出現し,MRIで中部胸髄中心部にT2強調画像高信号病変を認めた.血清IgG4高値,髄液蛋白上昇,抗AQP4抗体・抗MOG抗体・各種自己抗体陰性.ステロイド療法が奏効したが,1年後に延髄下端~上部頸髄に脊髄炎が再発した.脊髄炎はどちらも3椎体長に満たず,ステロイド治療が有効で,萎縮を残さなかった.近年,IgG4関連疾患(IgG4-related disease,以下IgG4-RDと略記)が疑われる脳脊髄炎が報告されている.今後,原因不明の脊髄炎とIgG4-RDとの関係を明らかにするために症例の蓄積と検討が必要である.
We report an 80-year-old man with IgG4-related pleuritis who had been treated with a low dose oral steroid for two years and developed recurrent myelitis. He was admitted to our hospital with gradually worsening numbness in the lower body and difficulty in walking due to mild weakness and loss of proprioception in the legs. T2-weighted MR images of the spinal cord showed a high signal intensity lesion, located centrally in the spinal cord at the Th2–4 spine levels. Laboratory data revealed an elevated serum IgG4 level and cerebrospinal fluid protein level. Anti-aquaporin 4 antibody, anti-myelin oligodendrocyte glycoprotein antibody and other autoantibodies were negative. He showed a good response to the administration of steroid pulse therapy with almost resolution of the neurological symptoms and MRI findings. He was followed with the maintenance therapy with a low dose oral steroid. After one year, he developed recurrence of myelitis in the lower end of the medulla oblongata and in the central to dorsal area at the C2 spine level. Each lesion of recurrent myelitis was located within 3 vertebral segments length and improved without focal spinal atrophy. Recently, IgG4-related disease (IgG4-RD)-associated inflammation involving brain parenchyma and spinal cord were reported. Further investigations are needed to elucidate the relationship between IgG4-RD and seronegative recurrent myelitis.
IgG4関連疾患(IgG4-related disease,以下IgG4-RDと略記)は,血清IgG4高値,組織への著明なIgG4陽性形質細胞浸潤と線維化,ステロイドに対する良好な反応性などを特徴とする全身性疾患である.多数の罹患臓器が報告され,神経系では肥厚性硬膜炎,下垂体炎が知られており,近年,末梢神経障害,さらに少数ながら脳実質炎や脊髄炎も報告されている.今回,我々はIgG4関連呼吸器疾患の治療中に再発性脊髄炎を発症した症例を経験したので,文献的考察を加えて報告する.
症例:80歳,男性
主訴:歩行障害,両下肢のしびれ
既往歴:高血圧,2型糖尿病,腰部脊柱管狭窄症.服用なし.
嗜好歴・家族歴:特記事項なし.
現病歴: 2017年5月,咳嗽,喘鳴を伴う呼吸困難が出現.右側胸水貯留,両側縦隔肺門リンパ節腫脹,胸膜肥厚が認められ,当院呼吸器外科・内科で精査された.PET-CTで右肺門縦隔,腸間膜に複数の集積像が認められたが,胸水細胞診や各種腫瘍マーカーで悪性所見なく,結核などの感染症も否定的だった.縦隔鏡による右壁側胸膜生検(Fig. 1)で,IgG4陽性形質細胞浸潤(10個/1視野以上,IgG4/IgG比 約50%)を認めた.花筵状線維化や血栓性静脈炎の所見は認めなかった.超音波内視鏡下穿刺吸引法による肺門リンパ節生検では腫瘍細胞,肉芽腫病変はなく,IgG4陽性形質細胞浸潤は僅かだった.血清IgG4 167 mg/dl(11.0~121.0 mg/dl),血清IgG 2,536 mg/dl(870~1,700 mg/dl)と高値であったので,経口プレドニゾロン20 mg/日を開始したところ,速やかに症状が改善し,胸水は消失した.治療経過も含めて,IgG4-RD(possible)と診断された1)2).気管支拡張薬とステロイド吸入薬による喘息治療を併用して,プレドニゾロンが漸減されたが,良好な全身状態で経過した.プレドニゾロン2.5 mg/日の時点の2018年1月に血清IgG4は448.0 mg/dlまで上昇したが,呼吸器症状の再燃はみられなかった.
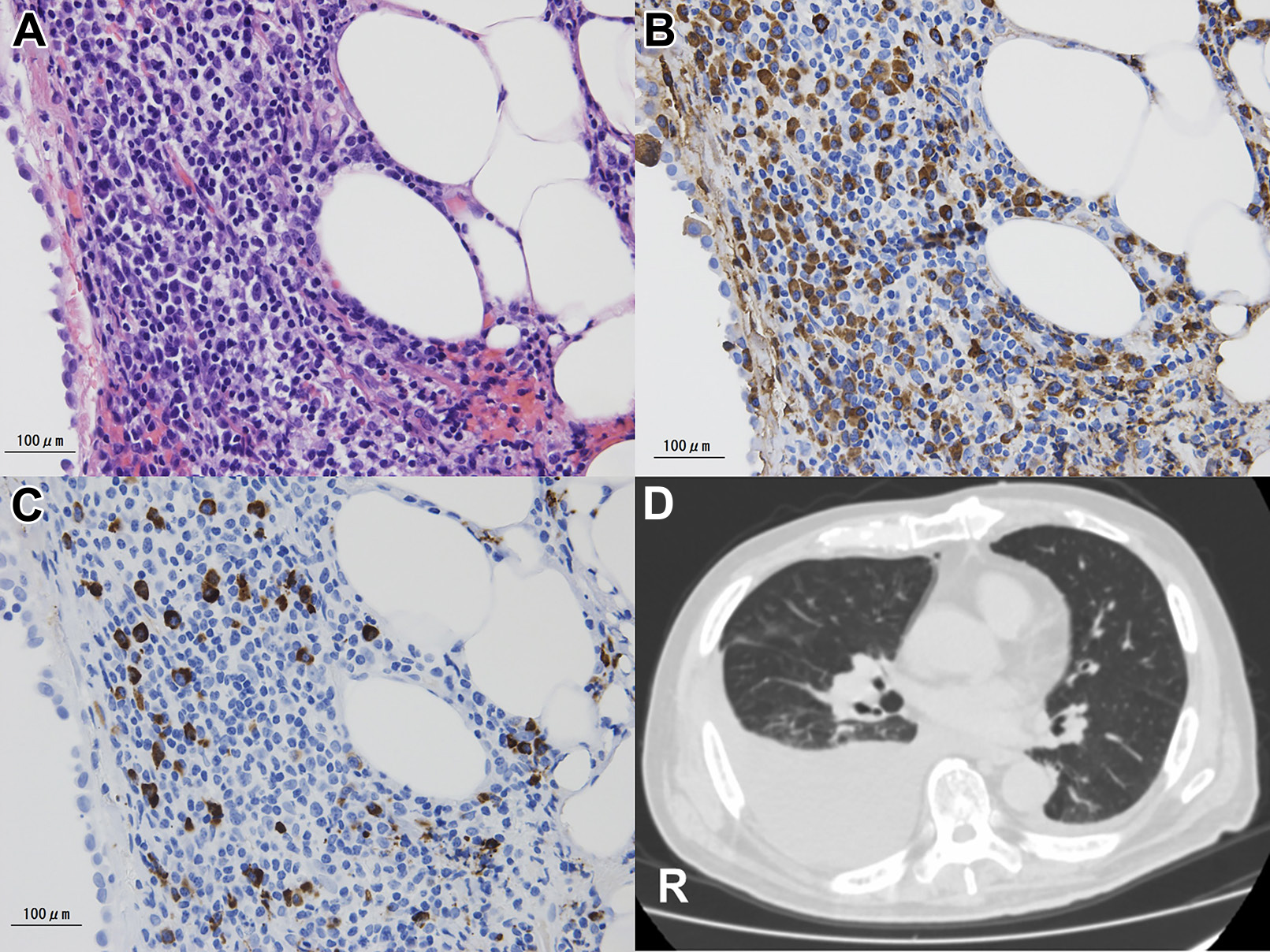
A. Histological findings of pleural biopsy specimen (Hematoxylin and Eosin staining) revealed packed infiltration of inflammatory cells. These cells consisted of mature small lymphocytes and plasma cells. Eosinophils are hardly seen. B. Most of lymphocytes and plasma cells are positive for IgG, immunohistochemically. C. Some of the IgG positive cells are positive for IgG4. The IgG4/IgG ratio was over 40%. D. CT image shows right-sided pleural effusion. A pleural biopsy was performed from the right pleura. (bar = 100 μm)
2019年6月下旬,右側腹部から腹部全体にしびれが出現し,次第に両下肢に広がり,脚がふらついてきたため,7月下旬に当科を受診した.
入院時現症:血圧115/66 mmHg,脈拍74/分・整,体温36.3°Cで,胸腹部に異常所見は認めなかった.皮疹なし.
神経学的所見:意識清明で見当識障害なし.脳神経系に異常所見なし.感覚検査では,Th6デルマトーム以下で表在感覚が鈍麻しており,右側腹部に持続的な鈍痛がみられた.振動覚は両側の手指で20秒と正常であったが,両足母趾で3秒と低下し,両足で関節位置覚が高度に低下していた.徒手筋力検査では,上肢筋力は正常,両下肢は腸腰筋3,膝屈・伸4,足関節背・底屈4だった.深部腱反射は正常範囲で,足底反射で両足母趾が軽度背屈した.膝踵試験で測定障害はみられなかった.歩行は開脚性のつたい歩き程度で,Romberg徴候陽性.自力排尿は可能だった.
検査所見:MRIでTh2~4レベルの脊髄中心部に,T2強調画像でおよそ2椎体長の高信号域が認められ,今回の責任病変と考えられた(Fig. 2A).また,延髄下端~C1レベルの頸髄中央から左側寄りにも淡い高信号病変が認められたが,これに対応する異常神経所見は認めなかった.大脳,下垂体,硬膜の異常所見はなかった.Gd造影は喘息の治療中であり行わなかった.髄液検査では,細胞数2個/μl,蛋白48 mg/dl,糖64 mg/dl(血糖115 mg/dl)と軽度に蛋白が上昇,IgG index 0.42,IgG4 6.0 mg/dl未満と正常だった.Oligoclonal IgG band,myelin basic proteinは測定しなかった.

A. STIR images (3.0 T) of the spinal MRI on July 2019. The sagittal image shows a hyper-intensity signal in the center of the thoracic spinal cord at the levels of Th2 to Th4. Another hyper-intensity signal is located at the levels from the lower medulla oblongata to the C1 spine. The axial image at the bottom of medulla oblongata shows a left-side dominant hyper-intensity lesion. B. STIR images on June 2020. The sagittal image shows a hyper-intensity signal from the bottom of medulla oblongata, and a large lesion occupying the central to dorsal area of the upper cervical cord. C. STIR images on September 2020. Both the sagittal and axial images show normalized lesions of the spinal cord at the upper cervical and thoracic levels.
一般血液検査では白血球数8,700/μl,好酸球15%と軽度の増多を認め,CRP 0.3 mg/dl,HbA1c 5.7%で,他の血算と生化学に異常はなかった.血清IgG 2,240 mg/dl,血清IgG4 170.0 mg/dlと高値だった.抗核抗体40倍以下,リウマトイド因子陰性,血清ACE 10.6 U/lと正常で,MPO-ANCA, PR3-ANCA,抗SS-A抗体,抗SS-B抗体,抗アクアポリン4(AQP4)抗体(EIA法),抗myelin-oligodendrocyte glycoprotein(MOG)抗体(Cell-based assay法,以下CBA法と略記)はいずれも陰性だった.胸腹部CTは正常だった.神経伝導検査,脛骨神経刺激での体性感覚誘発電位で明らかな異常はなかった.
入院後経過:自己免疫性脊髄炎として,day 2から,5日間のメチルプレドニゾロン1 g/日のパルス療法を2クールおこなった.パルス間の5日間とパルス後にプレドニゾロン(PSL)40 mg/日を投与し,day 5頃から歩行および感覚障害の改善がみられた.Day 17でほぼ脊髄炎発症前の歩行状態に回復して,PSL 30 mg/日で自宅退院した.退院時の血清IgGは1,318 mg/dlだった.
退院後,治療開始から4週経過での血清IgG4は30.2 mg/dl,血清IgG 890.0 mg/dlと正常化していた.MRIで頸髄と胸髄の病変は消失していた.PSLを漸減し,血糖上昇と軽度のステロイド精神病のため,2019年12月から2.5 mg/日を維持量としたが,呼吸器および神経系の症状再燃はなく,血清IgG4も30~50 mg/dlで経過した.
2020年6月初旬から四肢の運動・感覚障害が出現し,次第に進行し両手ともスプーンを持てなくなり,歩行不可能になったため,6月下旬に再診した.MRIで延髄下端からC2レベルの脊髄中心部にT2強調画像で高信号域と軽度腫脹が認められた(Fig. 2B).頸部以下で四肢遠位優位に表在感覚鈍麻,振動覚消失,関節位置覚著減.徒手筋力検査は四肢で3程度.起立保持不可能だが,両手を引けばすり足で移動できた.深部腱反射は正常範囲,下肢の病的反射陰性で,排尿は可能だった.血清IgG 1,417 mg/dlと正常範囲内であったが,2019年12月時の血清IgG 890.0 mg/dlより上昇していた.血清IgG4は35.5 mg/dl, CBA法による抗AQP4抗体は陰性であった.髄液検査で,細胞数0個/μl,蛋白48 mg/dl,oligoclonal IgG band陽性,myelin basic proteinは155 pg/mlと上昇していた.1回目同様に頭部MRIは異常所見を認めなかった.初回同様のステロイド治療を行ったところ,前回よりも反応は遅かったが,四肢の運動・感覚障害に改善がみられた.また,ステロイドパルス第1クール開始直後から数日間の持続性の吃逆が出現したが,嘔気・嘔吐はなかった.約1ヶ月の入院治療で上下肢の深部感覚は回復し,MRI画像も改善した(Fig. 2C).第4腰椎圧迫骨折を生じたための運動制限が加わったが,つたい歩き,箸で食事が可能となり,自宅退院した.退院後は10 mg/日のPSL維持で再発なく,半年後のMRIで脊髄萎縮は認めなかった.
本例は,IgG4-RDの包括診断基準1)および呼吸器疾患診断基準2),2019年のACR/EULARのIgG4-RD分類基準3)に照らして,疑診例(possible)に相当する.IgG4関連胸膜炎の寛解期に,再発性の脊髄炎を発症し,初回の脊髄炎では高IgG・高IgG4血症を伴った.再発時には血清IgG4値は正常範囲内であったが,IgG値の上昇傾向を認めており,IgG4-RDによる病態を疑った.
本例の再発性脊髄炎の鑑別疾患としては,好発年齢・性別は異なるが,多発性硬化症(multiple sclerosis,以下MSと略記),視神経脊髄炎スペクトラム障害(neuromyelitis optica spectrum disorder,以下NMOSDと略記),抗MOG抗体関連ミエロパチーが重要と考えられる.その他には,血清IgG4高値を呈することもある全身性エリテマトーデス,シェーグレン症候群などの膠原病,先行した呼吸器疾患とも関連するサルコイドーシスや悪性リンパ腫,また,神経ベーチェット病などが挙げられるが,いずれも胸膜炎発症時からの検査結果および治療経過から否定的である.また,自己免疫性glial fibrillary acidic protein(GFAP)アストロサイトパチーは髄膜炎や脳病変の合併がないことから鑑別した.
IgG4-RDの中枢神経系疾患としては肥厚性硬膜炎,下垂体炎が知られているが,近年,末梢神経障害の報告がみられるようになり,さらに肥厚性硬膜炎による圧迫もしくは炎症波及によらない,脳実質炎,偽腫瘍性腫瘤,脊髄炎の少数の報告がある4)~11).これらの中で,IgG4陽性形質細胞浸潤の病理所見が確認された報告は,脳生検がおこなわれたRegevらの1例のみである4).脊髄実質炎としては,Vakrakouらの報告があり,17歳女性で血清IgG4高値,無症候性の下垂体炎の合併,ステロイド治療の反応性からIgG4-RDが疑われた再発性脊髄炎の1例が記載されている.脊髄腫大を伴う脊髄中心部のC2~5レベル病変で,再発も同部位であった5).Linらは,47歳男性で,大脳・脳幹の多発病変と,Th6~7レベルから円錐部に連続する脊髄炎を呈し,腋窩リンパ節生検でIgG4陽性形質細胞浸潤を認めた症例を報告している6).両報告とも,抗AQP4抗体,抗MOG抗体は陰性であった.
本例の脊髄炎の画像的特徴については次のようであった.病変部位は,初発時,胸髄,再発時は延髄下端~上部頸髄で,いずれも約2椎体長の,脊髄中心部から後索にかけての病変で,脊髄腫脹はわずかだった.初発時にも延髄下端~C1レベルの無症候性病変が認められた.発症からの進行が炎症性疾患としてやや緩徐であった.症状は後索障害による下肢深部感覚障害が主体であった.再発時の症状極期に数日間の持続性吃逆が出現し,延髄最後野の症状と考えられた.ステロイド治療の効果は良好で,治療半年を経過した時点で病変部脊髄萎縮は認めなかった.本例の頸髄・胸髄病変の軸断像は,脊髄辺縁に片寄った小病変であることが多いMSよりも,脊髄中央ないしやや後方寄りに比較的大きい病変が生じるというNMOの特徴に似ていた.なお,抗MOG抗体関連ミエロパチーでは,矢状断で線状高信号,軸断像で灰白質の高信号がH型を呈することが特徴とされる12).IgG4-RDで脳軟膜炎を生じた報告があるが13)14),本例では造影MRIは行っておらず,脊髄表層軟膜炎からの波及の可能性は議論できないが,髄液所見や脊髄病変分布からは非典型的である.
本例の脊髄病変は,上記のようにNMOSDとの類似点があるが,2015年のNMOSD診断基準15)に照らすと,抗AQP4抗体陰性の本例では,延髄最後野病変・脳幹病変を認めたが,3椎体以上の長大な脊髄病変ではなく,局所的な脊髄萎縮を伴わないため,基準を満たさない.ただし,抗AQP4抗体陽性NMOSDでも短い脊髄炎を呈する場合は少なくない16).
IgG4-RDの病態について詳細は明らかになっておらず,現時点で確立した病因抗体は見つかっておらず,IgG4の直接的な役割についても不明である.中枢神経病変に関しては,Regevらの報告では,大脳皮質・皮質下で血管周囲腔のIgG4陽性形質細胞を含む炎症細胞浸潤が確認されており4),他に,血管炎の関与が考えられる症例がわずかに報告されている17)~19).他の自己免疫疾患の合併については,ANCA関連血管炎の合併は多く報告されるが,それ自体がIgG4-RD様の病態を伴うことがあるため合併とするには慎重を要する20).
IgG4-RDとNMOSDの合併例は検索した限りで報告はないが,先述のIgG4-RDに伴う脊髄炎の2報告は脊髄長大病変を呈していた.本例においても,IgG4-RDを生じる自己免疫賦活状態において,NMOに類似した脊髄炎を生じた可能性がある.脊髄病変の類似性からは,アストロサイトの補体介在性傷害であるNMO,オリゴデンドロサイトが標的とされる抗MOG抗体関連疾患などと共通する神経膠細胞障害に関する病態が存在するかもしれない.中枢神経実質病変は組織診断が困難なため,髄液GFAP濃度測定などによる病態の検討も重要と考えられる.
IgG4-RDの経過中に脊髄炎を生じる症例に関して,その臨床像や病態を明らかにするために今後の症例の蓄積が重要である.
謝辞:病理組織所見につきご教授いただいた,鈴鹿中央総合病院病理診断科 村田哲也先生に深謝いたします.
※著者全員に本論文に関連し,開示すべきCOI状態にある企業,組織,団体はいずれも有りません.