Abstract
β2 Adrenergic receptor (β2AR) is a kind of G-protein coupled receptors (GPCRs) which transduce a wide range of extracellular signals into intracellular messages responsible for the regulation of diverse cell functions. Because of their functional ubiquity, GPCR is one of the most important drug targets in pharmaceutical industry. Although recent crystallographic studies provided both the active and the inactive states of some families of GPCRs, the influence of lipid composition of bilayer membrane on their activation is still poorly understood. In this work, we address the influence of lipid composition on the structural stability of GPCR, performing molecular dynamics simulations of three kinds of states: apo-, and agonist epinephrine-, or antagonist alprenolol-bound β2AR. These three kinds of β2ARs were embedded in four types of lipid membranes: (i) pure palmitoyl-oleoyl-phosphatidyl-choline (POPC), (ii) POPC/cholesterol (CHL), (iii) POPC/CHL/GM1 (GM1 ganglioside), (iv) POPC/palmitoyl-oleoyl-phosphatidyl-ethanolamine (POPE)/CHL/sphingomyeline (SM). The side chains of Lys2676.29 and Asp3317.58 showed different conformations among the three states in all types of lipid membranes. The distances between Lys2676.29 and Asp3317.58 of apo- and alprenolol-bound β2ARs are smaller than that of the epinephrine-bound β2AR. In contrast, β2ARs in POPC/CHL bilayer were unstable in which the salt bridge; i.e., ionic lock, was not formed between Arg1313.50 and Glu2686.30. We have also examined the distribution of lipid molecules. A stable hydrophobic interaction between CHL and β2AR was observed at transmembrane helix5 in POPC/CHL/GM1 and POPC/POPE/CHL/SM membranes. These results suggest that the lipid composition strongly affects the conformation of GPCR and essentially concerns the GPCR activation.
G-Protein coupled receptors (GPCRs) are the largest class of versatile signaling molecules that are responsible for the signal transduction across cellular membranes in response to extracellular hormones or neurotransmitters, etc.1) Accordingly, GPCRs have been the major drug targets in pharmaceutical industry. The β2 adrenergic receptor (β2AR) is well studied member of GPCR families, which mediates the effects of catecholamine, epinephrine, and norepinephrine.2) β2AR plays an important regulatory role in a variety of cells and organs and is an important therapeutic target in the treatment of airway and cardiovascular disease.3) Like other GPCRs, the conformation of β2AR is known to vary between the inactive and the active states. The conformational change induced by the binding of extracellular ligands to β2AR results in the G-protein-mediated cell signaling.
The protein structures vary, depending on the inactive and the active signaling state of the receptor. The crystal structures of two GPCRs, β2AR and bovine rhodopsin, have recently been revealed both for the active state (agonist-bound) and the inactive state (antagonist-bound).4–6) The human β2AR is one of the first GPCRs to be identified through ligand binding, and is the first neurotransmitter receptor to be cloned.7) Its structure has been determined in complex with various kinds of ligands including inverse agonists, full agonists, and antagonists.8–10) In the previous computational studies, the inactive state of β2AR in complex with agonist or antagonist has been embedded in a lipid bilayer membrane composed of a single type of lipid molecule (palmitoyl-oleolyl-phosphatidyl-ethanolamine (POPE) bilayer or palmitoyl-oleoyl-phosphatidyl-choline (POPC) bilayer).6,11) Recent studies suggest that rafts, special kind of micro-domains that are enriched by cholesterol (CHL), play an essential role in the signaling by enabling the conformational change of GPCRs.12–16) In the present study, we analyze the inactive structure of β2AR without ligand (apo) or in complex with ligand (agonist epinephrine and antagonist alprenolol) embedded in the palmitoyl-oleoyl-phosphatidyl-choline (POPC) bilayer, cholesterol (CHL) containing POPC, GM1 ganglioside containing POPC/CHL, or the mixed membrane consisting of POPC, palmitoyl-oleoyl-phosphatidyl-ethanolamine (POPE), CHL, sphingomyeline (SM). The last one imitates the biological membranes, using a software program developed for modeling various types of lipid membranes.17) The mixed membrane, POPC/POPE/CHL/SM, was created referring the studies on the lipid composition of biological membrane.18–21) In erythrocyte, the lipid bilayer was composed of equal ratios by weight of cholesterol (CHL) and phospholipids.19) The three major phospholipids are POPC, POPE and SM.19,21) About 25% of the phospholipids is SM.21) Judging from these findings, 50% of the POPC/POPE/CHL/SM mixed membrane was set to CHL. SM was included at 12% in the mixed membrane. POPC and POPE were assigned to the rest 38%. Therefore, our simulation using the membrane model with the mixed lipid composition will be helpful to understand the structural perturbation of GPCRs in the biological membrane.
In the diverse intracellular signaling process, β2ARs constitute multiple receptor conformation in its active and inactive states. Biochemical and biophysical studies suggested that the rearrangement of helix3 and helix6 was involved in the activation process of rhodopsin and β2AR.22–24) Moreover the inactive rhodopsin structures demonstrated that highly conserved residue Arg1313.50 (superscripts refer to Ballesteros–Weinstein residue numbering scheme25)) at the end of intracellular part of helix3 formed a salt bridge; i.e., ionic lock, with acidic residue Glu2686.30 at the end of intracellular part of helix6.26–28) In contrast, an activated rhodopsin structure showed the break of the ionic lock between Arg1313.50 and Glu2686.30.22,23,29,30) Although the crystal structure of the inactive β2AR lacks the salt bridge between Arg1313.50 and Glu2686.30, several biochemical studies have suggested that an ionic lock between Arg1313.50 and Glu2686.30 is established in β2AR as well as other GPCRs.6,31–35) The disruption of ionic interaction between the highly conserved D(E)RY residue at helix3 and an acidic residue at helix6 is considered to promote the receptor activation and to be highly related to the conformational change of both rhodopsin and β2AR.22,31,35)
For understanding of the activation mechanism of β2AR embedded in a lipid bilayer membrane, in this work, we have performed molecular dynamics (MD) simulation by constructing computational models for ligand bound and unbound β2AR embedded in four types of lipid bilayer membranes; pure POPC, POPC/CHL, POPC/CHL/GM1 and POPC/POPE/SM/CHL, solvated with water molecules, and executed 100 ns MD simulations for every model system with applying periodic boundary conditions. We particularly address the effect of lipid composition of bilayer membrane on the ligand bound and unbound β2AR receptor. Several huge MD simulations previously characterized the dynamics of rhodopsin and/or β2AR and some simulation studies on β2AR mainly focused on the formation of ionic lock.6,36–40) Contrary to the huge simulations, a few reports14,15,41) have indicated that lipid composition modulates the activity of GPCRs. In MD simulations of two kinds of GPCRs of k-opioid receptor and rhodopsin in CHL-depleted and CHL-enriched model membranes, k-opioid receptor was suggested to functionalize better in CHL-enriched environments, while a relatively low concentration of CHL was favorable for rhodopsin.41) Therefore, GPCR function is closely linked to the composition of lipid membrane and CHL will highly affect the functional motion or conformational change of GPCRs.14,15,42–44) In the study of k-opioid receptor and rhodopsin, the DMPC/CHL bilayer was employed with setting the ratio of CHL 20%.41) In our POPC/CHL, POPC/CHL/GM1 and POPC/POPE/SM/CHL models, CHL was set to be about 50% of lipid molecules. Accordingly the amount of cholesterol is assumed to be sufficient to observe the β2AR activation.
In this study, MD simulations using the membrane models with different kind of lipid compositions showed the change in the ionic lock formation between Arg1313.50 and Glu2686.30 in apo-, epinephrine- and alprenolol-bound β2AR. We compared these results to the previous study performed using pure lipid bilayer POPE.6) The previous study showed that the ionic lock was formed in apo- and inverse agonist carazolol-bound β2AR. Our simulations with pure POPC and POPC/POPE/CHL/SM membranes were consistent with the previous study. Our simulation further suggested that the receptor in apo- and epinephrine- or alprenolol-bound form hardly maintained the ionic lock in POPC/CHL bilayer. This means that the composition of lipid membrane is essentially important for the activation of GPCR. One of the lipid components; CHL, has been observed to strongly interact with the receptor in the POPC/CHL/GM1 and POPC/POPE/CHL/SM mixed membrane. The present computational study will clarify the complex structure of GPCR and CHL and enhance the understanding of the influence on the receptor function.
Experimental
The initial structure of β2AR receptor for MD simulations was taken from an X-ray crystal structure of the carazolol-bound β2AR/T4-lysozyme fusion protein (code 2RH1) solved by Cherezov et al.45) The lysozyme and ligand carazolol were removed. The N-terminal 35 residues did not appear in the crystal structure. Hence, the missing residues were not included in our model of β2AR receptor. The protonation states of charged residues and histidines were determined on the basis of pKa values calculated by PROPKA.46) According to the PROPKA calculation results, the δ nitrogen atoms in His93, 172, 178, 269 and 296 were set to be protonated and the ε nitrogen atoms were deprotonated for those residues.
The full agonist epinephrine was manually docked into the β2AR and the neutral antagonist alprenolol was also docked to the β2AR, referring the crystal structure of ligand-bound β2AR (PDB code 3NYA).9) The chemical structures were subjected to geometrical optimization at the B3LYP/6-31G** level using Gaussian03 software. Atomic charges of the chemicals were determined by the restrained electrostatic potential (RESP) fitting procedure47) using the optimized structure. The force field parameters for epinephrine and alprenolol were determined by parameterizing fragment molecules and combining them according to the CHARMM parameterization methodology. The CHARMM force field for small organic molecules48) and the CHARMM27 force fields for lipids49) were employed for all simulation. All of the bilayer models were constructed using VMD ver. 1.850) and an in-house program named GLYMM which is a VMD plug-in to add a function of automatically making a heterogeneous lipid bilayer.17) Initially, the coordinates of all lipids were generated by the membrane tool of VMD, which provided the atom geometry of the liquid crystalline state of POPC lipids. Next, some of POPC molecules were converted into POPE, SM, CHL, or GM1 by GLYMM. The missing parameters were already created based on the analogy with available parameters.51) The POPC bilayer model consisted of 226 POPC molecules. The POPC/CHL bilayer consisted of 120 POPC and 112 CHL molecules (POPC/CHL=0.52 : 0.48). The POPC/CHL/GM1 bilayer consisted of 128 POPC, 104 CHL and 7 GM1 molecules (POPC/CHL/GM1=0.54 : 0.43 : 0.03). The POPC/POPE/SM/CHL bilayer consisted of 36 POPC, 58 POPE, 112 CHL and 26 SM molecules (POPC/POPE/CHL/SM=0.15 : 0.25 : 0.48 : 0.12). Hence, calculation models in this work provided various types of membrane environments in which β2AR receptor were placed. TIP3P water molecules and ions to neutralize the calculation cell were generated to solvate the complex of β2AR and lipid membrane, making a periodic boundary box of ca. 80 Å×90 Å×120 Å. Consequently, the total number of atoms was approximately 75000 in each model as shown in Fig. 1a.

MD simulations were carried out for every model using NAMD ver. 2.752) under the isothermal–isobaric (NPT) ensemble condition. The simulations were divided into four parts: minimization, heating, equilibration, and production runs. After the potential energy had been minimized, the model system was heated to 310 K with z coordinates of the head groups of the lipids restrained in order for the lipids to move only in the x and y directions. Gradually releasing the restraint, we equilibrated the system at a constant temperature of 310 K and a constant pressure of 1 atm and then carried out production runs under no restraint condition. The simulation times for equilibration and production runs were 10 ns and 100 ns, respectively. Non-bonded interaction terms were computed with a cutoff distance of 12 Å, where a switching distance of 10 Å was applied to make the non-bonded interaction zero at the cutoff distance smoothly. An integration time step was 2 fs. A periodic boundary condition was applied to all directions of the calculation cell in a similar manner to the previous work,53) and the particle mesh Ewald method was employed to compute the long-distance non-bonded interaction. CHARMM27 force field49) was adopted for all the atoms except for ligand chemical.
The root mean square deviation (RMSD) measurement was performed, using the ptraj module of AMBER11.54) In this analysis, only the coordinates for C, O, and N atoms of the main chain of receptor were used, because the flexibility of the side chain is large. The trajectories acquired from the 100 ns MD simulation were fit to the snapshot structure after the 10 ns equilibration run as a reference. The average structure was calculated using the trajectories for the last 20 ns, because the RMSD showed small values for the last 20 ns in MD simulation.
Fluctuation of main chain atoms for the respective residues of the receptor was examined using the trajectories for 100 ns of the MD simulation. Fluctuation was evaluated by the B-factor values55,56) calculated by the ptraj module of AMBER11 without including side chain. The structure after equilibration was used as the reference, and the trajectories were fit to this reference with respect to C, O, and N atoms in the main chain to eliminate the movement due to the rotation and the transversal motions.
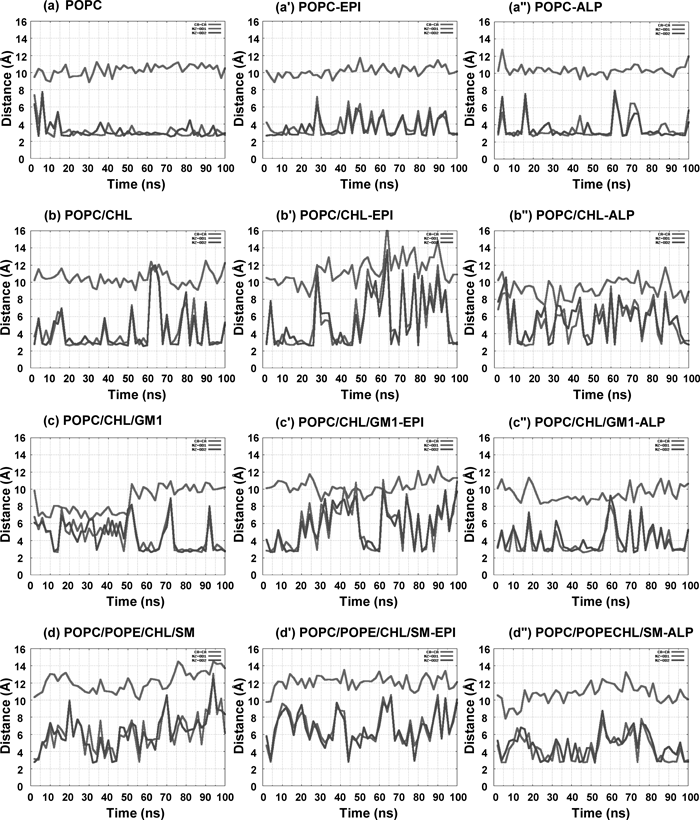
The Cα–Cα and N–O distances shown in Fig. 4 were computed from the snapshot structures of the 100 ns MD simulation. We focused on two major conformational changes in which one is the inactive; ionic lock is formed, and another is the active state; ionic lock is broken, as shown in Fig. 1c. The N–O and Cα–Cα distances regarding to the ionic lock were measured to examine whether the ionic lock is formed or broken between Arg1313.50 and Glu2686.30. Another distance between Lys2676.29 of intracellular end of helix6 and Asp3317.58 of intracellular end of helix7 was also measured with respect to Cα–Cα and N–O distances as shown in Fig. 1d.
The distribution of lipid molecules around β2AR was measured with respect to their distance and direction and was visualized using gnuplot. Each coordinate of lipid molecule in the respective trajectories was rotated and superimposed so that β2AR were fitted to the reference one.
Results
We carried out 12 patterns of 100 ns MD simulations for β2AR embedded in pure and/or mixed membrane bilayers as shown in Table 1. The crystal structure of β2AR; PDB code: 2RH1, shown in Fig. 1b was used for the receptor. For the ligand-free, apo form, the simulations are labeled as a, b, c and d. An agonist epinephrine (Fig. 1e) was docked into the binding pocket manually referring to the literature,57,58) and the simulations are labeled as a′, b′, c′ and d′. An antagonist alprenolol (Fig. 1f) was placed at the binding site, referring the crystal structure; PDB code: 3NYA,9) and the simulations are labeled as a″, b″, c″ and d″. The simulations using pure POPC bilayer were labeled as a, a′ and a″. POPC bilayer containing CHL was used in simulations b, b′ and b″. POPC, CHL and GM1-ganglioside containing membrane was used in simulations c, c′ and c″. The mixed membrane consisting of POPC, POPE, CHL and SM was used in simulations d, d′ and d″.
Table 1. Calculation Pattern of the MD Simulations
| Simulation | Components of lipid bilayer | Protein | Ligand | Simulation time (ns) |
|---|
| a | POPC | β2AR | — | 100 |
| a′ | POPC | β2AR | Epinephrine | 100 |
| a″ | POPC | β2AR | Alprenolol | 100 |
| b | POPC/CHL | β2AR | — | 100 |
| b′ | POPC/CHL | β2AR | Epinephrine | 100 |
| b″ | POPC/CHL | β2AR | Alprenolol | 100 |
| c | POPC/CHL/GM1 | β2AR | — | 100 |
| c′ | POPC/CHL/GM1 | β2AR | Epinephrine | 100 |
| c″ | POPC/CHL/GM1 | β2AR | Alprenolol | 100 |
| d | POPC/POPE/SSM/CHL | β2AR | — | 100 |
| d′ | POPC/POPE/SSM/CHL | β2AR | Epinephrine | 100 |
| d″ | POPC/POPE/SSM/CHL | β2AR | Alprenolol | 100 |
Fluctuation of protein structure is closely related to the stability of protein. Flexibility of protein was highly correlated with protein functions.59) B-factor analysis presents the deviation of atom positions from their average points. The calculated B-factor values of β2AR for the respective models are shown in Fig. S1 of Supplementary Information. A comparison among simulations a, a′ and a″ suggested that, in POPC bilayer, the flexibility of protein residues in apo form was higher than those in the presence of epinephrine and alprenolol, and simulation a″ gave the smallest fluctuation (Fig. 2a). Therefore, alprenolol stabilized the β2AR structure. The flexibility of protein residues for POPC/CHL bilayer model was significantly decreased compared to that of pure POPC bilayer (Fig. 2b). Simulations for POPC/CHL/GM1 and mixed POPC/POPE/CHL/SM bilayers showed further small flexibility compared with the pure POPC and POPC/CHL bilayer (Figs. 2c, d). In the presence of alprenolol, simulations always showed the small flexibility for protein residues than other simulations. These results suggest that the lipid molecule strongly influences the stability of the residues and alprenolol also affects the flexibility of the receptor.

Figures 3a and b show the superimposition of the average structures of simulations for epinephrine-bound and alprenolol-bound β2ARs to the apo-β2AR in the POPC/POPE/CHL/SM mixed membrane (simulations d, d′, d″). The averaged structures were obtained from the last 20 ns MD simulations (Fig. S2) and pymol software60) were employed to superimpose the structures with respect to main chain atoms of β2AR. β2AR is bound to Gα protein with making hydrogen-bond network between them, in which Thr682.39, Asp1303.49 and Arg1313.50 of β2AR are strongly involved in the binding.61) The comparison of β2AR structures among simulations d, d' and d″ indicated that the Gα binding site kept the almost similar conformation as shown by blue mesh in Figs. 3a and b. To analyze the interactions between ligands and β2AR receptor in all the models (Figs. S3, S4), we evaluated the hydrogen-bond network connecting ligand and β2AR. In the presence of epinephrine (Fig. 3c, Figs. S3, S4), epinephrine interacted with Ser2035.42, Ser2075.46 and with Asp1133.32 of β2AR. The movement of helix5, which has also been investigated in the recently published crystal structures of β2AR,4,10,61) is important for forming the hydrogen-bond network between ligand and receptor. In case of alprenolol (Fig. 3d, Figs. S3, S4), it interacted with Asp1133.32 but no interaction with Ser2035.42 and Ser2075.46. Hence, the inverse agonist and/or antagonist will be not involved in the movement of helix5, which is compatible with the previous computational study.61) The interactions between ligand (epinephrine or alprenolol) and β2AR (Figs. 3c, d) indicated that a stable interaction maintained in the simulation.

It is important to examine the activation of β2AR in terms of the contribution of the intracellular amino acid residues to the formation of ionic lock (Fig. 4). The ionic lock was continuously formed in pure POPC and POPC/POPE/CHL/SM mixed membrane containing apo-, and epinephrine- or alprenolol-bound β2ARs. In the simulations a, a′, a″ and d, d′, d″, β2AR maintained a formation of ionic lock and the intracellular ends of helix3 and helix6 made a close network with each other (Fig. 4). Arg1313.50 and Glu2686.30 interacted with each other via salt bridge during the MD simulation. In a β2AR crystal structure; PDB code: 2RH1, Cα–Cα and N–O distances of Arg1313.50 and Glu2686.30 were 11.2 Å and 9.9 Å respectively, but typical distances of the inactive rhodopsin structures were ca. 8.8 Å and ca. 2.8 Å. The Cα–Cα and N–O distances were ca. 9.5 Å and ca. 3.0 Å throughout the 100 ns simulations in a–a″ and d–d″ (Fig. 4). Both of these simulations showed that the ionic lock was formed and helix3 and helix6 were close to each other. The N–O distance in Fig. 4 shows two pairs of lines, one pair represents NH1 to OE1 and OE2 distances and another one represents NH2 to OE1 and OE2 distances. The distances of the two lines in the respective pairs were observed to synchronize with each other. The side chain atoms of Arg1313.50 and Glu2686.30 fluctuated during the simulation while they always maintained the formation of ionic lock. No noticeable difference was observed among apo-, epinephrine- and alprenolol-bound β2AR.
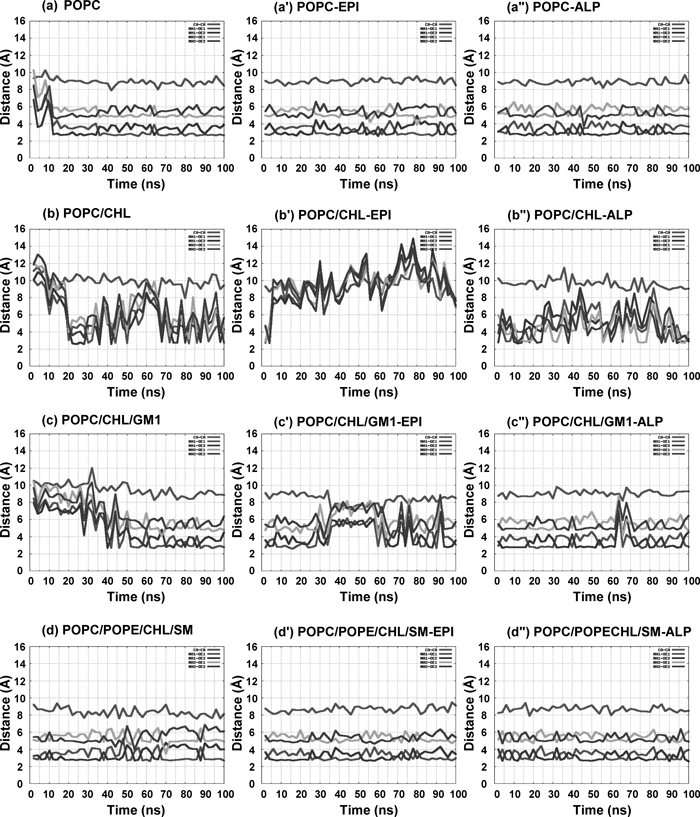
The apo-, and epinephrine- or alprenolol-bound β2AR in POPC/CHL bilayer in simulations b, b′ and b″ showed the break of the ionic lock (Fig. 4). The ionic lock was broken frequently in these three simulations. The N–O distances show no clear separation in four lines. The displacement of the side chain atoms of Arg1313.50 and Glu2686.30 are large compared to the simulations with other type of lipid membrane. The apo-, and epinephrine- or alprenolol-bound β2ARs in the POPC/CHL/GM1 bilayer in simulations c, c′ and c″ showed the instability of the ionic lock (Fig. 4). The ionic lock distance was frequently enlarged compared with simulations a–a″ and d–d″. The present findings suggest that the ionic lock distance depends on the lipid compositions.
Characteristic Conformational Changes in the Side Chain ResiduesThe conformational change of side chain will affect on Gα protein bound with β2AR. Hence we analyzed the conformation of the side chains at the intracellular part of β2AR, especially for three residues Thr682.39, Asp1303.49 and Arg1313.50 that are involved in the binding to Gα protein. The conformations of these three residues are similar among all the models. The distances between two of these three residues are represented in Figs. S5 and S6 of Supplementary Information. The Cα–Cα and N–O distances give the almost similar kinds of results in apo-, and epinephrine- or alprenolol-bound β2AR in all models. We observed the Cα–Cα and N–O distances between Thr682.39 and Asp1303.49 in all simulations. The distances were ca. 7.5 Å and 3–6 Å in a–a″, c–c″ and d–d″ (Fig. S5). In contrast, the distances were ca. 9.5 Å and 5–8.5 Å in the CHL bilayer simulations in b–b″. We also evaluated the Cα–Cα and N–O distances between Thr682.39 and Arg1313.50 in all the simulations. The distances were ca. 8.5 Å and 4 Å in a–a″, c–c″ and d–d″ (Fig. S6), but these distances were 9–10.5 Å and 4–6 Å in CHL containing membrane in b–b″. These changes in distance are similar to Fig. 4. In contrast, Lys2676.29 and Asp3317.58 showed a different conformational change among all the models (Fig. 5). The distance between Lys2676.29 and Asp3317.58 was relatively short in apo- and alprenolol-bound β2AR in all models whereas the distances became long in the epinephrine-bound β2AR. Judging from these N–O distances, we concluded that the interaction was stably maintained between Lys2676.29 and Asp3317.58 in a and a″. In contrast, this interaction was often broken in a′ for the epinephrine-bound β2AR. The N–O distance between Lys2676.29 and Asp3317.58 was occasionally maintained in b and b″, c and c″, d and d″ and the interaction was almost broken in b′, c′ and d′. Therefore, the distance between Lys2676.29 and Asp3317.58 will be one of the important indices that varies depending on the presence and the absence of antagonist.
The distribution of lipid molecules were analyzed using the trajectories acquired from the last 30 ns MD simulation (Fig. S7). The lipid molecules, which are located near around β2AR, showed sharp distributions, whereas distributions became broad for the lipid molecules that are apart from β2AR. The sharpness in distribution will be due to the interaction between lipid molecules and β2AR. The lipid molecule, CHL, is found to be distributed heterogeneously in all the models (Fig. S7). Superimpositions of the distribution of lipid molecules in apo- and epinephrine- or alprenolol-β2AR for the respective membrane models are shown in Fig. S8. In case of POPC/POPE/CHL/SM mixed membrane, two CHLs marked by red circle always stay at the same position during last 30 ns MD simulations (Fig. 6a). This position is the location between the helix4 and helix5 of β2AR. Figure 6b shows the top view of CHL distributions around β2AR and the CHLs colored red are bound to β2AR. CHL has a highly hydrophobic part. In the upper and lower leaflets of the lipid membrane, two CHLs mainly interact with the non-polar hydrophobic residues of Ala1985.37, Ala2025.41, Val2065.45, Pro1684.60, Ile2145.53, Val1263.45 and Val1293.48 (Fig. 6c). The CHL in the upper leaflet interacts with Ala1985.37, Ala2025.41, and Val2065.45 and Pro1684.60. The CHL in the lower leaflet interacts with Val2065.45, Ile2145.53 and Val1263.45, Val1293.48. Hence the hydrophobic interaction tightly connects CHL molecule to helix5 of β2AR.

Discussion
Protein flexibility is closely related to the function of a protein. We have analyzed the residue fluctuation by B-factor analysis. The comparison of B-factor among the simulation models indicated that the flexibility of protein residues in apo form is higher than those in the presence of epinephrine and alprenolol. Protein residues give the smallest fluctuation in the presence of alprenolol, which suggests that alprenolol stabilizes the β2AR structure. Goetz et al. carried out MD simulation on the agonist isoprenaline- or inverse agonist carazolol-bound β2AR.61) Their comparison of B-factor between the isoprenaline- and the carazolol-bound β2ARs demonstrated that the residue fluctuation was reduced in the presence of carazolol, which is consistent with our results (Fig. S1, Fig. 2).
Some previous studies have revealed that the binding modes of epinephrine and alprenolol to β2AR were different.9,10,62–64) In computational approaches, Simpson et al. carried out MD simulation on the epinephrine-bound β2AR.62) In their calculation the hydrogen-bond interactions with epinephrine were observed at Ser2035.42, Ser2075.46 and Asp1133.32, which is compatible with our results (Fig. 3c, Figs. S3, S4). An X-ray crystal analysis suggested the interaction of Ser2035.42 and Ser2075.46 with a ligand.10) In many computational studies on the structure of the agonist-bound and the inverse agonist/antagonist-bound β2ARs, an agonist was found to make hydrogen-bonds with receptor through the residues of Ser2035.42, Ser2075.46 and Asp1133.32.63,64) In contrast, the inverse agonist/antagonist bound to β2AR was shown to make the hydrogen-bond network with Asp1133.32 and not to establish the hydrogen-bonds with Ser2035.42 and Ser2075.46,9) which is also consistent with our current findings (Fig. 3d, Figs. S3, S4).
The ionic lock formation of GPCR in lipid membrane has already been studied in simulation and suggested to play an important role in the activation of GPCR.6,65) About 70% of GPCRs of the rhodopsin family contain the E(D)RY(W) motif on helix3. Arg1353.50 of the conserved motif makes an intrahelical salt bridge to the neighboring Glu1343.49. Arg1353.50 also interacts with Glu2476.30 strongly. Further, Glu2496.32 interacts with the backbone NH of Lys3117.58 at the kink from helix7 to the small cytoplasmic helix8. During the activation of rhodopsin, the interhelical interactions between Arg1353.50 and Glu2476.30, and between Glu2496.32 and Lys3117.58 were observed to be broken.65) The disruption of the intrahelical salt bridge between Glu1343.49 and Arg1353.50 at the cytoplasmic terminus of helix3 was proposed to be a key factor for the transition to the active conformation.6,65)
An important difference between the crystallographic structures of rhodopsin26–28) and β2AR5,8,45,66) bound with the partial inverse agonist carazolol and timolol is the disruption of the ionic lock. The β2AR structures lack the interhelical interactions between homologous residues Arg1313.50 and Glu2686.30, and between Lys2706.32 and Asp3317.58. Dror et al. carried out MD simulation on a carazolol-bound β2AR embedded in pure POPE lipid bilayer.6) They showed that the Cα–Cα distance was below 9.5 Å for 91% of the simulation time. According to their results, the ionic lock was stably maintained between the backbones of Arg1313.50 and Glu2686.30 for the inverse agonist bound β2AR. This finding is consistent with our results using the pure POPC bilayer. But the intrahelical salt bridge between two adjacent residues Asp1303.49 and Arg1313.50 appear to be preserved and it stabilizes the inactivated form of the receptor. Furthermore, intracellular ends of helices 3 and 6 are substantially far apart from each other compared with those in rhodopsin.6) The disruption of the ionic lock was previously suggested to be the crucial step for the GPCR activation. Some mutagenesis studies indicated that the mutations of the residues related to the ionic lock in β2AR and those of homologous residues in rhodopsin-family GPCRs resulted in a continuous receptor activation.31–33,65) In spite of the above suggestion, the ionic lock may be not only major factor in GPCR activation because β2AR has the broken ionic lock in the crystallographic structures.
In the inactive state of β2AR, Lys2706.32 and Asp3317.58 were suggested not to interact with each other in contrast to the interaction between the homologous residues; Glu2496.32 and Lys3117.58, in the inactive state of rhodopsin.65) In our results we found that another residue, Lys2676.29 kept an adequate distance to Asp3317.58 in apo- and alprenolol-bound β2AR in all calculation models. In the epinephrine-bound β2AR structures, this distance became noticeably larger in all models, which suggests the presence of a different kind of salt bridge in the inactive form of β2AR.
The transmembrane receptors are in contact with the membrane lipid bilayer, which suggests that membrane has an important influence on receptor structure and function.67) Hence, it is necessary to analyze the effect of lipid composition on the structural stability of the receptor. As well as other membrane proteins,14) the optimal function of GPCR will depend on the lipid environment. The significance of specific lipids in the function of GPCR has been demonstrated in a variety of studies.68,69) For example, the presence of phosphatidylethanolamine (PE) was shown to modulate the activation of GPCR.68) In addition, the presence of phospholipids with unsaturated fatty acids in the membrane is required for efficient signaling of rhodopsin.69) Similar to other lipid molecules, CHL is also required for the optimal function of membrane proteins.67) The lipid-receptor interactions, especially CHL-receptor interaction, is involved in the modification of conformation and function of several GPCR family proteins.15) CHL is distributed heterogeneously in domains or pools of biological and artificial membranes70) and believed to be important for the maintenance of membrane structure. CHL modulates the conformation and the function of membrane proteins by a direct molecular interaction with the receptor71) or due to the alteration of the membrane physical properties induced by the presence of CHL.67,72,73) For example, CHL is directly bound to the sterol regulatory element binding protein cleavage activating protein (SCAP), which is a large polytopic membrane protein, thereby CHL induces a conformational change in SCAP.
The CHL-receptor interactions have extensively been studied in the case of rhodopsin. Due to the increase of the amount of CHL, rhodopsin shifted itself to the inactive conformation.74) Molecular dynamic simulation with rhodopsin in a membrane containing CHL and unsaturated phospholipids suggested the direct interaction between rhodopsin and CHL75) and hence the enhancement of the stability of the receptor in the membrane. Further, CHL is required for keeping a high binding affinity between ligand and receptor. For example, the efficiency of ligand binding to the cholecystokinin receptor was reported to be reduced by the lowering of the concentration of CHL in membrane.76) The importance of CHL in membrane for the ligand binding was also suggested in subtype 2 galanin (GalR2) and oxytocin receptor.77,78) Moreover, CHL-receptor interaction was also found in case of β2AR. A recent X-ray crystal analysis suggested that CHL molecules were tightly bound to β2AR45) and CHL seemed to be an important component for crystallization of the receptor. These findings indicate that CHL composes a specific interaction with β2AR and promotes the enzymatic functions for ligand binding, receptor-G-protein interaction, and/or downstream signal transduction.79) Hanson et al. provided the crystal structure of β2AR bound with both CHL and a partial inverse agonist timolol and showed that CHL was bound at a shallow surface groove formed by segments of helices 1, 2, 3, and 4.8) CHL was shown to enhance the thermal stability of β2AR, by forming many intermolecular interactions through van der Waals interactions or hydrogen bonding to β2AR helix. For example, Tyr702.41 makes the van der Waals interaction with ring A of CHL and a hydrogen bond to Arg1514.43 in β2AR.8) A recent study proposed a characteristic amino acid sequence, named as cholesterol recognition/interaction amino acid consensus (CRAC) motif, which is the dominant region interacting with CHL and observed in three representative GPCRs of rhodopsin, β2AR and serotonin1A receptor. β2AR presents CRAC motifs in transmembrane helix5; residues 231–221, and helix7; residues 324–328.80) In our molecular dynamics study, CHL binding was observed in all the β2AR in apo- and epinephrine- or alprenolol-bound form and the contribution of CHL for β2AR stability was suggested. In our simulations except for pure POPC model, CHL appeared to be distributed heterogeneously in the membranes and a specific CHL-binding to the helices 3, 4 and 5 in β2AR was observed in the simulations c–d.″ CHL molecules mainly interacted with the hydrophobic residues such as Ala1985.37, Ala2025.41, Val2065.45 and Ile2145.53, with its principle molecular axis almost parallel to the membrane normal direction. The change of the distances between CHL and these residues were monitored for the last 30 ns of MD simulation (Fig. S9). The fluctuation of these distances was small during the simulation, which suggested that the hydrophobic interaction was stable. In order to evaluate the interaction from the quantitative viewpoint, the shape-complementarity between CHL and β2AR was measured. The shape-complementarity was calculated to be was 0.68. This value is higher than those of the antigen-antibody interfaces, while slightly lower than those of the interfaces between proteases and their inhibitors.81) Therefore, the molecular shape of CHL is concluded to be well compatible with the helix4 and helix5 region of β2AR.
Conclusion
The influence of the lipid composition on the structural conformation of one kind of GPCR protein, β2AR, was investigated by performing molecular dynamics simulations. Four types of lipid membranes, pure POPC, POPC/CHL, POPC/CHL/GM1 and POPC/POPE/CHL/SM containing three kinds of β2AR; apo- and the agonist epinephrine- or the antagonist alprenolol-bound form, were employed for 100 ns simulation and the computational results were compared. This work presented the following findings. (1) The stability of the ionic lock formation between Arg1313.50 and Glu2686.30 varies depending on the difference of the lipid component of membrane. (2) The side chain interaction between Lys2676.29 and Asp3317.58 became strong in apo- and the alprenolol-bound β2AR, compared with the epinephrine-bound β2AR in pure POPC membrane. (3) A stable hydrophobic interaction was observed between CHL and the receptor at the transmembrane helix5 in POPC/CHL/GM1 and POPC/POPE/CHL/SM membranes. CHL is specifically bound at the position between helix4 and helix5 in β2AR.
Acknowledgment
Calculations were performed at Research Center for Computational Science, Okazaki, Japan and at Information Technology Center of the University of Tokyo. This work was partly supported by a Grant for Scientific Research C from Japan Society for the Promotion of Science.
References
- 1) Audet M., Bouvier M., Nat. Chem. Biol., 4, 397–403 (2008).
- 2) Lefkowitz R. J., Biochim. Biophys. Acta, 1768, 748–755 (2007).
- 3) Jones S. M., Hiller F. C., Jacobi S. E., Foreman S. K., Pittman L. M., Cornett L. E., BMC Pharmacol., 3, 15 (2003).
- 4) Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K., Nature (London), 477, 549–555 (2011).
- 5) Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., Kobilka B. K., Science, 318, 1266–1273 (2007).
- 6) Dror R. O., Arlow D. H., Borhani D. W., Jensen M. Ø., Piana S., Shaw D. E., Proc. Natl. Acad. Sci. U.S.A., 106, 4689–4694 (2009).
- 7) Dixon R. A., Kobilka B. K., Strader D. J., Benovic J. L., Dohlman H. G., Frielle T., Bolanowski M. A., Bennett C. D., Rands E., Diehl R. E., Mumford R. A., Slater E. E., Sigal I. S., Caron M. G., Lefkowitz R. J., Strader C. D., Nature (London), 321, 75–79 (1986).
- 8) Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., Stevens R. C., Structure, 16, 897–905 (2008).
- 9) Wacker D., Fenalti G., Brown M. A., Katritch V., Abagyan R., Cherezov V., Stevens R. C., J. Am. Chem. Soc., 132, 11443–11445 (2010).
- 10) Rosenbaum D. M., Zhang C., Lyons J. A., Holl R., Aragao D., Arlow D. H., Rasmussen S. G., Choi H. J., Devree B. T., Sunahara R. K., Chae P. S., Gellman S. H., Dror R. O., Shaw D. E., Weis W. I., Caffrey M., Gmeiner P., Kobilka B. K., Nature (London), 469, 236–240 (2011).
- 11) Dror R. O., Arlow D. H., Maragakis P., Mildorf T. J., Pan A. C., Xu H., Borhani D. W., Shaw D. E., Proc. Natl. Acad. Sci. U.S.A., 108, 18684–18689 (2011).
- 12) Chini B., Parenti M., J. Mol. Endocrinol., 32, 325–338 (2004).
- 13) Pike L. J., J. Lipid Res., 44, 655–667 (2003).
- 14) Pucadyil T. J., Chattopadhyay A., Prog. Lipid Res., 45, 295–333 (2006).
- 15) Burger K., Gimpl G., Fahrenholz F., Cell. Mol. Life Sci., 57, 1577–1592 (2000).
- 16) Lingwood D., Simons K., Science, 327, 46–50 (2010).
- 17) Mori K., Hata M., Neya S., Hoshino T., Chem. Bio. Info. J., 4, 15–26 (2004).
- 18) Madison K. C., Wertz P. W., Strauss J. S., Downing D. T., J. Invest. Dermatol., 87, 253–259 (1986).
- 19) Ingraham L. M., Burns C. P., Boxer L. A., Baehner R. L., Haak R. A., J. Cell Biol., 89, 510–516 (1981).
- 20) Brügger B., Glass B., Haberkant P., Leibrecht I., Wieland F. T., Kräusslich H. G., Proc. Natl. Acad. Sci. U.S.A., 103, 2641–2646 (2006).
- 21) Winterbourn C. C., Batt R. D., Biochim. Biophys. Acta, 202, 1–8 (1970).
- 22) Farrens D. L., Altenbach C., Yang K., Hubbell W. L., Khorana H. G., Science, 274, 768–770 (1996).
- 23) Sheikh S. P., Zvyaga T. A., Lichtarge O., Sakmar T. P., Bourne H. R., Nature (London), 383, 347–350 (1996).
- 24) Jensen A. D., Guarnieri F., Rasmussen S. G., Asmar F., Ballesteros J. A., Gether U., J. Biol. Chem., 276, 9279–9290 (2001).
- 25) Ballesteros J. A., Weinstein H., Methods Neurosci., 25, 366–428 (1995).
- 26) Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M., Science, 289, 739–745 (2000).
- 27) Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V., J. Mol. Biol., 342, 571–583 (2004).
- 28) Li J., Edwards P. C., Burghammer M., Villa C., Schertler G. F. X., J. Mol. Biol., 343, 1409–1438 (2004).
- 29) Altenbach C., Kusnetzow A. K., Ernst O. P., Hofmann K. P., Hubbell W. L., Proc. Natl. Acad. Sci. U.S.A., 105, 7439–7444 (2008).
- 30) Scheerer P., Park J. H., Hildebrand P. W., Kim Y. J., Krauss N., Choe H. W., Hofmann K. P., Ernst O. P., Nature (London), 455, 497–502 (2008).
- 31) Ballesteros J. A., Jensen A. D., Liapakis G., Rasmussen S. G., Shi L., Gether U., Javitch J. A., J. Biol. Chem., 276, 29171–29177 (2001).
- 32) Greasley P. J., Fanelli F., Rossier O., Abuin L., Cotecchia S., Mol. Pharmacol., 61, 1025–1032 (2002).
- 33) Angelova K., Fanelli F., Puett D., J. Biol. Chem., 277, 32202–32213 (2002).
- 34) Shapiro D. A., Kristiansen K., Weiner D. M., Kroeze W. K., Roth B. L., J. Biol. Chem., 277, 11441–11449 (2002).
- 35) Yao X., Parnot C., Deupi X., Ratnala V. R., Swaminath G., Farrens D., Kobilka B., Nat. Chem. Biol., 2, 417–422 (2006).
- 36) West G. M., Chien E. Y., Katritch V., Gatchalian J., Chalmers M. J., Stevens R. C., Griffin P. R., Structure, 19, 1424–1432 (2011).
- 37) Tikhonova I. G., Best R. B., Engel S., Gershengorn M. C., Hummer G., Costanzi S., J. Am. Chem. Soc., 130, 10141–10149 (2008).
- 38) Spijker P., Vaidehi N., Freddolino P. L., Hilbers P. A. J., Goddard W. A. 3rd, Proc. Natl. Acad. Sci. U.S.A., 103, 4882–4887 (2006).
- 39) Huber T., Menon S., Sakmar T. P., Biochemistry, 47, 11013–11023 (2008).
- 40) Sato Y., Hata M., Neya S., Hoshino T., J. Phys. Chem. B, 110, 22804–22812 (2006).
- 41) Khelashvili G., Mondal S., Andersen O. S., Weinstein H., J. Phys. Chem. B, 114, 12046–12057 (2010).
- 42) Visiers I., Ballesteros J. A., Weinstein H., Methods Enzymol., 343, 329–371 (2002).
- 43) Filizola M., Weinstein H., Curr. Opin. Drug Discov. Devel., 8, 577–584 (2005).
- 44) Weinstein H., AAPS J., 7, E871–E884 (2005).
- 45) Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C., Science, 318, 1258–1265 (2007).
- 46) Li H., Robertson A. D., Jensen J. H., Proteins, 61, 704–721 (2005).
- 47) Gilson M. K., Sharp K. A., Honig B. H., J. Comput. Chem., 9, 327–335 (1988).
- 48) Momany F. A., Rone R., J. Comput. Chem., 13, 888–900 (1992).
- 49) Feller S. E., MacKerell A. D., J. Phys. Chem. B, 104, 7510–7515 (2000).
- 50) Humphrey W., Dalke A., Schulten K., J. Mol. Graph., 14, 33–38 (1996).
- 51) Mori K., Mahmood M. I., Neya S., Matsuzaki K., Hoshino T., J. Phys. Chem. B, 116, 5111–5121 (2012).
- 52) Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K., J. Comput. Chem., 26, 1781–1802 (2005).
- 53) Imai Y., Liu X., Yamagishi J., Mori K., Neya S., Hoshino T., J. Mol. Graph. Model., 29, 461–469 (2010).
- 54) Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Crowley M., Walker R. C., Zhang W., Merz K. M., Wang B., Hayik S., Roitberg A., Seabra G., Kolossvary I., Wong K. F., Paesani F., Vanicek J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Yang L., Tan C., Mongan J., Hornak V., Cui G., Mathews D. H., Seetin M. G., Sagui C., Babin V., Kollman P. A., “AMBER11 University of California,” San Francisco (2010).
- 55) Zoete V., Michielin O., Karplus M., J. Mol. Biol., 315, 21–52 (2002).
- 56) Mori K., Hata M., Neya S., Hoshino T., J. Am. Chem. Soc., 127, 15127–15137 (2005).
- 57) Katritch V., Reynolds K. A., Cherezov V., Hanson M. A., Roth C. B., Yeager M., Abagyan R., J. Mol. Recognit., 22, 307–318 (2009).
- 58) de Graaf C., Rognan D., J. Med. Chem., 51, 4978–4985 (2008).
- 59) Daniel R. M., Dunn R. V., Finney J. L., Smith J. C., Annu. Rev. Biophys. Biomol. Struct., 32, 69–92 (2003).
- 60) DeLano W. L., “Pymol software,” The DeLano Scientific, San Carlos, CA, U.S.A. (2002).
- 61) Goetz A., Lanig H., Gmeiner P., Clark T., J. Mol. Biol., 414, 611–623 (2011).
- 62) Simpson L. M., Wall I. D., Blaney F. E., Reynolds C. A., Proteins, 79, 1441–1457 (2011).
- 63) Wolf S., Böckmann M., Höweler U., Schlitter J., Gerwert K., FEBS Lett., 582, 3335–3342 (2008).
- 64) Liapakis G., Ballesteros J. A., Papachristou S., Chan W. C., Chen X., Javitch J. A., J. Biol. Chem., 275, 37779–37788 (2000).
- 65) Vogel R., Mahalingam M., Lüdeke S., Huber T., Siebert F., Sakmar T. P., J. Mol. Biol., 380, 648–655 (2008).
- 66) Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K., Nature (London), 450, 383–387 (2007).
- 67) Lee A. G., Biochim. Biophys. Acta, 1666, 62–87 (2004).
- 68) Alves I. D., Salgado G. F., Salamon Z., Brown M. F., Tollin G., Hruby V. J., Biophys. J., 88, 198–210 (2005).
- 69) Mitchell D. C., Niu S. L., Litman B. J., J. Pediatr., 143 (Suppl.), S80–S86 (2003).
- 70) Schroeder F., Woodford J. K., Kavecansky J., Wood W. G., Joiner C., Mol. Membr. Biol., 12, 113–119 (1995).
- 71) Gimpl G., Burger K., Fahrenholz F., Trends Biochem. Sci., 27, 596–599 (2002).
- 72) Yeagle P. L., Biochim. Biophys. Acta, 822, 267–287 (1985).
- 73) Ohvo-Rekilä H., Ramstedt B., Leppimäki P., Slotte J. P., Prog. Lipid Res., 41, 66–97 (2002).
- 74) Albert A. D., Young J. E., Yeagle P. L., Biochim. Biophys. Acta, 1285, 47–55 (1996).
- 75) Pitman M. C., Grossfield A., Suits F., Feller S. E., J. Am. Chem. Soc., 127, 4576–4577 (2005).
- 76) Harikumar K. G., Puri V., Singh R. D., Hanada K., Pagano R. E., Miller L. J., J. Biol. Chem., 280, 2176–2185 (2005).
- 77) Pang L., Graziano M., Wang S., Biochemistry, 38, 12003–12011 (1999).
- 78) Gimpl G., Burger K., Politowska E., Ciarkowski J., Fahrenholz F., Exp. Physiol., 85 (Spec. No.), 41S–49S (2000).
- 79) Ben-Arie N., Gileadi C., Schramm M., Eur. J. Biochem., 176, 649–654 (1988).
- 80) Jafurulla M., Tiwari S., Chattopadhyay A., Biochem. Biophys. Res. Commun., 404, 569–573 (2011).
- 81) Lawrence M. C., Colman P. M., J. Mol. Biol., 234, 946–950 (1993).