Abstract
Furonaphthoquinones are promising skeletons for anticancer drug molecules. In particular, methoxylated furonaphthoquinones are characteristic constituents of Tabebuia plants. In this research, we synthesized the furonaphthoquinones by effective one-pot cascade reactions of 3-phenyliodonio-1,2,4-trioxo-1,2,3,4-tetrahydronaphthalenides with 3-butyn-2-ol in the presence of palladium and cuprous catalysts via Sonogashira coupling and intramolecular cyclization. Furthermore, we demonstrated that the synthetic furonaphthoquinones showed moderate cytotoxicity against human leukemia U937 and HL-60 cells. Our work highlights the importance of furonaphthoquinones as antileukemic agents.
Tabebuia plants belong to the Bignoniaceae family, which is commonly found in North and South America. The plants have been used for the treatment of cancer as folk medicines, Pau d’Arco, Ipé Roxo, Taheebo, and Lapacho.1) In 1982, 2-(1-hydroxyethyl)naphtho[2,3-b]furan-4,9-dione FN0-ol and 2-acetylnaphtho[2,3-b]furan-4,9-dione FN0-one were isolated from Tabebuia cassinoides. Both these furonaphthoquinones showed significant activity against human oral epidermoid carcinoma KB cells.2) Subsequently, FN0-one was investigated as an anticancer agent in vitro, and it was found that this quinone shows moderate cytotoxicity against various cancer cell lines, including human oral squamous carcinoma HSC-2, HSC-3, HSC-4, human prostate carcinoma DU-145, human melanoma C8161, human lung adenocarcinoma A549, and human epithelial carcinoma HeLa cells.3–6) Previous phytochemical investigations of Tabebuia plants resulted in the isolation and characterization of a variety of furonaphthoquinones bearing a methoxy group in the benzene ring.7–9) Thus, these methoxylated furonaphthoquinones were confirmed to be characteristic constituents of Tabebuia plants.
As described above, furonaphthoquinones are considered interesting natural compounds in biochemical and pharmacological fields. Herein, we report the detailed synthesis of these furonaphthoquinones and their antileukemic activity.
Results and Discussion
Some of naturally occurring furonaphthoquinones bear a 1-hydroxyethyl group at the C2-position. Kobayashi et al. reported a synthesis method of furonaphthoquinones based on sequential coupling/ring closure reactions.10) However, the reaction with intact 3-butyn-2-ol was unsuccessful. Thus, we attempted to improve their method, with the aim of making it useful for the synthesis of a wide variety of the compounds having a hydroxy group. 2-(1-Hydroxyethyl)furonaphthoquinones such as FN0-ol are envisaged to be formed through the in situ generation of iodonaphthoquinones from phenyliodonium ylides in the presence of cuprous oxide (Cu2O), followed by Sonogashira coupling with acetylene and subsequent intramolecular cyclization. The phenyliodonium ylide 3-phenyliodonio-1,2,4-trioxo-1,2,3,4-tetrahydronaphthalenide IY0, which would serve as a suitable precursor for the subsequent reactions, was easily prepared from lawsone NQ0 using (diacetoxyiodo)benzene, without partition and purification by chromatography.11) Because the reaction did not progress even after 24 h in the presence of 3 mol% palladium(II) acetate (Pd(OAc)2) and 2.0 eq 3-butyn-2-ol in pyridine at room temperature (Table 1, entry 1), the amounts of Pd(OAc)2 and acetylene were increased to 10 mol% and 10 eq, respectively. Under these conditions, the desired product was obtained in 28% yield (entry 2). However, under the above mentioned conditions, the yield did not improve even after the reagents were changed to Pd(OAc)2/CuI or tetrakis(triphenylphosphine)palladium (Pd(PPh3)4)/Cu2O (entries 3, 4). At 80°C, the yield improved to 40% (entry 5) but remained unaffected by an increase in the amount of Cu2O (entry 6). For further improvement of the yield, we employed additives such as cuprous bromide (CuBr), CuI, silver(I) oxide (Ag2O), 2,2′-bipyridyl (2,2′-bpy), and 1,4-diazabicyclo[2.2.2]octane (DABCO).12–15) The best result, i.e., 56% yield, was obtained when the reaction was carried out for 4 h using CuBr (entry 7). A longer reaction time led to decomposition of the product. It was considered that CuBr activates the formation of cuprous acetylide and that its pyridine complex plays a crucial role in the intramolecular cyclization step.16,17)
Table 1. Optimization of Reaction Conditions for the Synthesis of 2-(1-Hydroxyethyl)furonaphthoquinone
FN0-ol from Phenyliodonium Ylide
IY0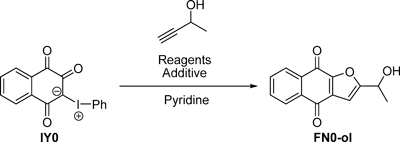 |
|---|
| Entry | Reagents | Additive | Acetylene (eq) | Temp. | Time | Yield (%) |
|---|
| 1 | 3 mol% Pd(OAc)2+Cu2O (1.0 eq) | — | 2.0 | rt | 24 h | Trace |
| 2 | 10 mol% Pd(OAc)2+Cu2O (1.0 eq) | — | 10 | rt | 24 h | 28 |
| 3 | 10 mol% Pd(OAc)2+CuI (1.0 eq) | — | 10 | rt | 24 h | Trace |
| 4 | 10 mol% Pd(PPh3)4+Cu2O (1.0 eq) | — | 10 | rt | 24 h | 9 |
| 5 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | — | 10 | 80°C | 4 h | 40 |
| 6 | 10 mol% Pd(OAc)2+Cu2O (3.0 eq) | — | 10 | 80°C | 4 h | 37 |
| 7 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | CuBr (1.0 eq) | 10 | 80°C | 4 h | 56 |
| 8 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | CuBr (1.0 eq) | 10 | 80°C | 12 h | 49 |
| 9 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | CuI (1.0 eq) | 10 | 80°C | 4 h | 46 |
| 10 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | Ag2O (1.0 eq) | 10 | 80°C | 4 h | 14 |
| 11 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | 2,2′-bpy (1.0 eq) | 10 | 80°C | 4 h | 41 |
| 12 | 10 mol% Pd(OAc)2+Cu2O (2.0 eq) | DABCO (1.0 eq) | 10 | 80°C | 4 h | 32 |
Then, we synthesized naphthoquinones having a methoxy group in the benzene ring. 2-Hydroxy-5-, 6-, and 7-methoxynaphthoquinones NQ5–7 were prepared by the reaction of methoxy-1-tetralones with potassium t-butoxide in t-butanol in O2 atmosphere.18) 2-Hydroxy-8-methoxynaphthoquinone NQ8 was synthesized from juglone in three steps19) (Chart 1). Naphthoquinones NQ0 and NQ5–8 were smoothly converted to the phenyliodium ylides IY0 and IY5–8, respectively. It is noteworthy that the optimized reactions with the ylides proceeded successfully to give the desired 2-(1-hydroxyethyl)furonaphthoquinones FN0-ol and FN5–8-ols (Chart 2). In the case of 5- or 8-methoxylated compounds, the yields were poor. It was suggested that an oxygenated group at the 5- or 8-position could enhance the electronegativity of the neighboring carbonyl oxygen.19) This involvement might affect the reactivity of a hydroxy group. Then, pyridine chlorochromate (PCC) oxidation of FN0-ol and FN5–8-ols to the corresponding 2-acetylfuronaphthoquinones FN0-one and FN5–8-ones in good yields could be achieved. Their NMR, MS, and melting point data were compared with the compounds in the literature.7–9) This strategy is expected to be applicable for the relatively simple synthesis of several other furonaphthoquinones as well.
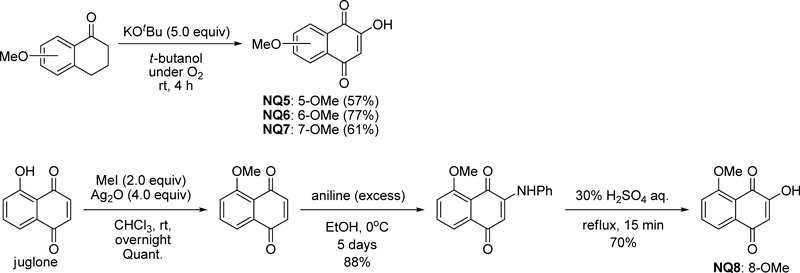
Chart 1. Preparation of Methoxylated Naphthoquinones

Chart 2. Synthesis of Furonaphthoquinones
Molecules that preferentially exhibit cytotoxicity against neoplastic cells may be used for developing drugs that are effective against cancer in humans. Quinone anticancer agents include anthraquinones [e.g., adriamycin (ADM)], benzoquinones [e.g., mitomycin-C (MMC)], and naphthoquinones [e.g., lapachone]. A variety of furonaphthoquinones with different substituent patterns showed potential for biological activity with various pharmacological actions and sometimes enhanced the anticancer activity.20) In our research, we assumed that incorporating an additional methoxy moiety into the furonaphthoquinones would improve their efficacy. As the next step, we evaluated the synthetic furonaphthoquinones for in vitro cytotoxicity against U937 and HL-60 cells using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay method.21) All the compounds inhibited cell proliferation in a concentration-dependent manner. Among the compounds, FN0-ol and FN0-one showed prominent effects with IC50 values of 1.4 and 0.97 µm in U937 cells (Table 2). On the other hand, the effects of FN5-ol, FN8-ol, and FN8-one, which have a methoxy group at the 5- or 8-position, were relatively weak. In HL-60 cells, FN0-one, FN6-one, and FN7-one also exhibited the potent effects. The IC50 values of these three furonaphthoquinones were 0.58, 0.87, and 0.59 µm, respectively (Table 2); in contrast, cisplatin, which is a famous anticancer drug, had an IC50 of 1.9 µm toward HL-60 cells.22) Interestingly, 2-acetylfuronaphthoquinones (ketones) tended to show stronger cytotoxicity than did 2-(1-hydroxyethyl)furonaphthoquinones (alcohols) in the both cells. These furonaphthoquinones have the potential to decrease glutathione (GSH) content, with an increase in glutathione disulfide (GSSG) formation. Naphthoquinone derivative-dependent depletion of GSH may be due to several mechanisms including direct conjugation23) or redox cycle-generated reactive oxygen species (ROS) such as superoxide anion and hydrogen peroxide, which can be detoxified by glutathione peroxidase with concomitant formation of oxidized glutathione.24)
Table 2. IC
50 Values of Various Furonaphthoquinones against U937 and HL-60 Cells
| IC50 (µm) |
|---|
| U937 | HL-60 |
|---|
| FN0-ol | 1.4 | 3.4 |
| FN0-one | 0.97 | 0.58 |
| FN5-ol | 4.8 | 3.4 |
| FN5-one | 3.3 | 2.1 |
| FN6-ol | 3.3 | 2.4 |
| FN6-one | 3.0 | 0.87 |
| FN7-ol | 4.1 | 4.5 |
| FN7-one | 3.7 | 0.59 |
| FN8-ol | 4.9 | 8.2 |
| FN8-one | 4.0 | 4.6 |
In conclusion, we have developed a direct one-pot cascade reaction via Sonogashira coupling and intramolecular cyclization for the synthesis of furonaphthoquinones. Pd(OAc)2 and cuprous catalysts play significant roles in the conversion. Overall, this methodology seems to be useful for the synthesis of naturally occurring furonaphthoquinones. Through the proposed synthetic route, several methoxylated furonaphthoquinones isolated from Tabebuia plants have been prepared. Additionally, we suggest that furonaphthoquinones would act as in vitro antileukemic agents against U937 and HL-60 cells. These first-generation scaffolds can be promising stepping stones to the discovery of more potent furonaphthoquinones that effectively inhibit leukemia cell proliferation.
Experimental
General chemistry methods, synthesis procedures, spectral data, and bioassay methods are given in Supplemental information.
References
- 1) Abbott B. J., Hartwell J. L., Leiter J., Perdue R. E. Jr., Schepartz S. A., Cancer Res., 27B, 190–199 (1967).
- 2) Rao M. M., Kingston D. G. I., J. Nat. Prod., 45, 600–604 (1982).
- 3) Hirai K. I., Koyama J., Pan J., Simamura E., Shimada H., Yamori T., Sato S., Tagahara K., Tsuruo T., Cancer Detect. Prev., 23, 539–550 (1999).
- 4) Eyong K. O., Kumar P. S., Kuete V., Folefoc G. N., Nkengfack E. A., Baskaran S., Bioorg. Med. Chem. Lett., 18, 5387–5390 (2008).
- 5) Rieber M., Rieber M. S., Cancer Biol. Ther., 7, 1206–1211 (2008).
- 6) Takano A., Hashimoto K., Ogawa M., Koyanagi J., Kurihara T., Wakabayashi H., Kikuchi H., Nakamura Y., Motohashi N., Sakagami H., Yamamoto K., Tanaka A., Anticancer Res., 29, 455–464 (2009).
- 7) Zani C. L., de Oliveira A. B., de Oliveira G. G., Phytochemistry, 30, 2379–2381 (1991).
- 8) de Oliveira A. B., Raslan D. S., de Oliveira G. G., Maia J. G. S., Phytochemistry, 34, 1409–1412 (1993).
- 9) Diaz F., Medina J. D., J. Nat. Prod., 59, 423–424 (1996).
- 10) Kobayashi K., Uneda T., Kawakita M., Morikawa O., Konishi H., Tetrahedron Lett., 38, 837–840 (1997).
- 11) Hatzgrigoriou E., Spyroudis S., Varvoglis A., Liebigs Ann. Chem., 1989, 167–170 (1989).
- 12) Mori A., Kawashima J., Shimada T., Suguro M., Hirabayashi K., Nishihara Y., Org. Lett., 2, 2935–2937 (2000).
- 13) Li P., Wang L., Wang M., You F., Eur. J. Org. Chem., 5946–5951 (2008).
- 14) Keddie D. J., Fairfull-Smith K. E., Bottle S. E., Org. Biomol. Chem., 6, 3135–3143 (2008).
- 15) Yamashita M., Ueda K., Sakaguchi K., Iida A., Tetrahedron Lett., 52, 4665–4670 (2011).
- 16) Zhou L., Shi Y., Xiao Q., Liu Y., Ye F., Zhang Y., Wang J., Org. Lett., 13, 968–971 (2011).
- 17) Huang M., Feng Y., Wu Y., Tetrahedron, 68, 376–381 (2012).
- 18) Kasturi T. R., Arunachalam T., Can. J. Chem., 44, 1086–1089 (1966).
- 19) MacLeod J. W., Thomson R. H., J. Org. Chem., 25, 36–42 (1960).
- 20) Ogawa M., Koyanagi J., Sugaya A., Tsuda T., Ohguchi H., Nakayama K., Yamamoto K., Tanaka A., Biosci. Biotechnol. Biochem., 70, 1009–1012 (2006).
- 21) Efdi M., Ninomiya M., Suryani E., Tanaka K., Ibrahim S., Watanabe K., Koketsu M., Bioorg. Med. Chem. Lett., 22, 4242–4245 (2012).
- 22) Akihisa T., Kikuchi T., Nagai H., Ishii K., Tabata K., Suzuki T., J. Oleo Sci., 60, 71–77 (2011).
- 23) Miller M. G., Rodgers A., Cohen G. M., Biochem. Pharmacol., 35, 1177–1184 (1986).
- 24) Salas C., Tapia R. A., Ciudad K., Armstrong V., Orellana M., Kemmerling U., Ferreira J., Maya J. D., Morello A., Bioorg. Med. Chem., 16, 668–674 (2008).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/FT1.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)