Review
Total Synthesis of Biologically Active Natural Products Based on Highly Selective Synthetic Methodologies
2014 年 62 巻 11 号 p. 1045-1061
詳細
2014 年 62 巻 11 号 p. 1045-1061
Total syntheses of structurally and biologically intriguing natural products relying on new synthetic methodologies are described. This article features cinchona alkaloid-catalyzed asymmetric Morita–Baylis–Hillman reactions, heterocycle syntheses based on rhodium-catalyzed C–H amination and indium-catalyzed Conia-ene reactions, and their utilization for the syntheses of the phoslactomycin family of antibiotics, glutamate receptor agonists and antagonists, and alkaloids with characteristic highly substituted pyrrolidinone core structures.
Natural products that selectively interact with specific receptors and enzymes are of great importance as new drug leads or probes for exploring significant biological events. For the supply of such molecules, efficient total synthesis is required, especially when their availability from natural resources is low or ethically unsound. Natural products are not only valuable scaffolds for drug discovery but also challenging synthetic targets that provide an opportunity to develop new synthetic methods based on innovative, rational strategies. To achieve the synthesis of a target molecule, we often need to develop and optimize new methodology, which extends to more generalized synthetic methods beyond the specific setting of the natural product, and in this way total synthesis leads to the development of new chemistry. Our research therefore focuses on the development of new methodologies useful for the construction of complex molecules and the total synthesis of structurally and biologically intriguing natural products. This review presents our representative achievements from such synthetic efforts.
The Morita–Baylis–Hillman (MBH) reaction is essentially a three-component reaction involving the coupling of the α-position of an activated alkene such as acrylate with a carbon electrophile such as aldehyde under nucleophilic amine or phosphine catalysis and produces synthetically useful multifunctional products. When an imine is used as a carbon electrophile, the reaction is called the aza-MBH reaction (Chart 1). The MBH reaction including the aza-version is regarded as one of the most promising carbon–carbon bond-forming reactions in terms of synthetic utility, atom economy, and operational simplicity. The growing importance of this reaction is evidenced by a quantum increase in the number of publications dealing with it during the past 15 years. However, the major problems associated with this reaction are its slow reaction rate and difficulty in achieving a high level of asymmetric induction.1–6)

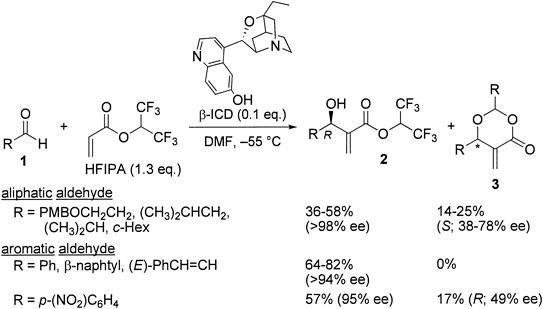
In 1999, we achieved the first breakthrough in the catalytic asymmetric MBH reaction employing β-isocupreidine (β-ICD) as a chiral bifunctional catalyst and 1,1,1,3,3,3-hexafluoroisopropyl acrylate (HFIPA) as an activated alkene (β-ICD–HFIPA method)7) (Chart 2). The reactions were carried out using 0.1 eq. of β-ICD and 1.3 eq. of HFIPA in N,N-dimethylformamide (DMF) at −55°C. In the case of aliphatic aldehydes, (R)-esters 2 were obtained with excellent enantioselectivity, although the yields were moderate due to the production of (S)-dioxanones 3 with opposite chirality and irregular ee values. In the reaction of aromatic aldehydes, high R-selectivity was again observed. Interestingly, the corresponding dioxanones 3 were not produced in these cases except for p-nitrobenzaldehyde, which gave the R-enriched dioxanone 3. These results indicate that aromatic aldehydes reacted with HFIPA with excellent enantioselectivity under β-ICD–HFIPA conditions in contrast to aliphatic aldehydes.
It was confirmed that both the cage-like tricyclic structure and the phenolic OH of β-ICD as well as the branched structure of HFIPA are essential for obtaining a high level of asymmetric induction as well as rate acceleration7,8) (Chart 3). On the basis of these findings and a computational study as well as Aggarwal’s proton transfer mechanism,10) we proposed a reaction mechanism governed by stereocontrol of the aldol step and proton transfer step from two energetically favored zwitterionic intermediates 5 and 6 stabilized by hydrogen bonding.9) This mechanism as well as Aggarwal’s rationale10) suggests that in order to develop an effective asymmetric MBH reaction we need to design a catalytic system that has the capability of controlling both the aldol and proton transfer steps. After our discovery of this β-ICD–HFIPA method, a number of sophisticated catalytic systems were developed.1–6)

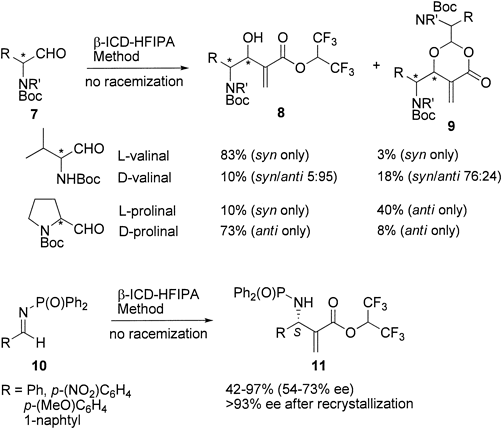
This β-ICD–HFIPA method is applicable to chiral α-amino aldehydes 7 that afford the synthetically useful α-methylene-β-hydroxy-γ-amino acid esters 811) (Chart 4). It was found that β-ICD-catalyzed reactions of N-Boc-α-amino aldehydes 7 with HFIPA proceeded without racemization to give enantiomerically pure esters 8 and exhibited a marked match–mismatch relationship. For example, L-valinal matched with the enantio-preference (R-selectivity) of β-ICD, affording excellent syn-selectivity and high chemical yields. On the other hand, D-valinal mismatched with β-ICD, showing a lack of diastereoselectivity and poor reactivity. Interestingly, L- and D-prolinals showed the opposite match–mismatch relationship to that observed for the acyclic substrates: D-prolinal rather than L-prolinal matched well with β-ICD, resulting in excellent anti-selectivity and good yield. Since the major problems associated with the MBH reaction of a chiral α-amino aldehyde are the racemization of the substrate and difficulty in securing high diastereoselectivity,12,13) our β-ICD–HFIPA method exhibits a marked advantage in this reaction.
We also examined the aza-MBH reaction of imine-type substrates, which allows us to construct valuable α-methylene-β-amino acid derivatives in optically active form.14) As can be seen in Chart 4, β-ICD-catalyzed reactions of aromatic N-(diphenylphosphinoyl)imines 10 gave N-diphenylphosphinoyl-α-methylene-β-amino acid esters 11 in moderate to good yields and enantioselectivities. Fortunately, the products were crystalline and the optical purity was enriched to >93% ee by simple recrystallization. Interestingly, the absolute configuration of the aza-MBH products was always S-enriched in contrast to that observed for the MBH reaction of aldehydes.
The β-ICD–HFIPA method has remarkable advantages due to the high enantioselectivity, broad applicability, and availability of both β-ICD16) and HFIPA as described above. However, one serious drawback is that this method cannot be applied to the synthesis of products with opposite absolute configurations because the required enantiomer of β-ICD is not easily available via synthesis.17–20) To solve this problem, we have long been studying the synthesis of a pseudoenantiomer of β-ICD21,22) and recently developed α-isocupreine (α-ICPN), which is available in excellent yield in one step from quinine and can be utilized as an enantiocomplementary catalyst of β-ICD in various β-ICD-catalyzed MBH reactions15) (Chart 5). We found that when quinine is heated in 28 eq. of CF3SO3H at 50°C for 24 h and then at 80°C for 15 h, successive rearrangements23–25) accompanied by demethylation occur to generate α-ICPN. The nuclear Overhauser effect spectroscopy (NOESY) spectrum and X-ray crystallography26) of α-ICPN indicates that it takes an anti conformation where the nucleophilic nitrogen faces toward the phenolic hydroxy group like β-ICD. As expected from this structural feature, α-ICPN showed the opposite enantioselectivity to that observed for the β-ICD-catalyzed reaction in the reactions of various aldehydes with HFIPA. In the case of aromatic aldehydes, esters 2 were obtained with high S-selectivity in the range of 82–93% ee in moderate to good yields. On the other hand, the reactions of aliphatic aldehydes yielded S-enriched esters 2 with high enantioselectivity up to 93% ee, although the isolated yields were moderate because of the production of dioxanones 3. This reaction tendency is the same as that observed for β-ICD-catalyzed reactions.7,8)

To demonstrate the utility of α-ICPN, we examined aza-MBH reactions of aromatic imines 12 with β-naphthyl acrylate (13) employing catalyst 1427) derived from α-ICPN and β-naphthol under the dual catalysis conditions developed by Masson and colleagues28–30) As summarized in Chart 6, the reactions of 12 and 13 produced aza-MBH adducts 15 with high R-selectivity in the range of 80% ee to 96% ee, which is opposite to that observed in the reactions catalyzed by the corresponding β-ICD-derived catalyst.28) The observed R-selectivity can be explained by assuming zwitterionic intermediates 1628) stabilized by hydrogen bonding, from which a six-membered proton transfer followed by E1cb elimination of the catalyst takes place to give R-aza-MBH product 15.

Since we developed the above-mentioned cinchona alkaoid-catalyzed asymmetric MBH reaction, we have demonstrated the synthetic utility of this reaction via syntheses of biologically intriguing natural products.31–33) In this review article, the enantio- and stereoselective syntheses of the phoslactomycin family of antibiotics are presented. Phoslactomycins A–F and I, produced by the soil bacteria species Streptomyces, constitute the phoslactomycin family of antifungal and antitumor antibiotics together with phosphazomycins C1 and C2, leustroducsins A–C and H, PD113,2714, and fostriecin.34) These compounds are highly potent selective inhibitors of protein serine/threonine phosphatase 2A, which is proposed to be responsible for their antitumor activity.34–37) Due to their intriguing molecular architectures and potential as lead compounds for anticancer drugs as well as their importance as a biological tool, this family of compounds has attracted much attention in the chemical and biological research communities. Thus, there have been a number of synthetic studies including formal and total syntheses of phoslactomycin A38) and B,39–42) leustroducsin B,43–47) fostriecin,47–49) and PD113,271.50) However, in spite of their similarity, previously reported synthetic methods are not of general utility in preparing all members of this family.
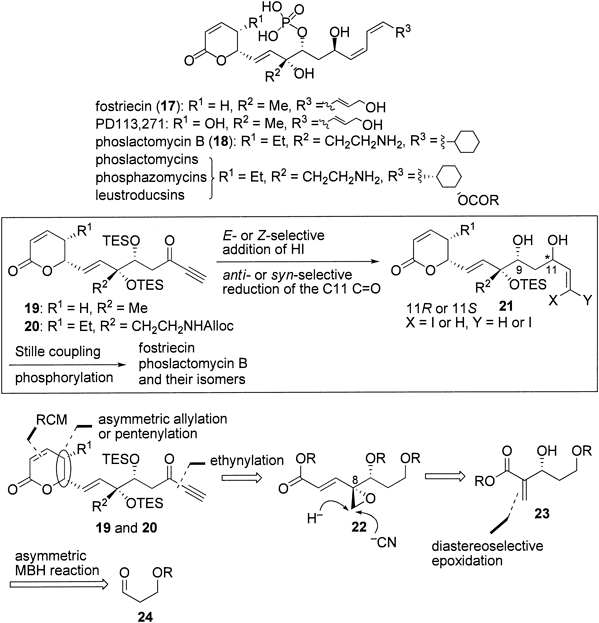
Previously, we demonstrated41,51,52) that ynones 19 and 20 effectively served as pivotal intermediates for the synthesis of 17 and 18 as well as their various isomers via Z- or E-selective conjugate addition of hydroiodic acid, C9-hydroxy-directed anti- or syn-selective reduction of the C11-carbonyl group, Stille coupling, and phosphorylation as major transformations (Chart 7). From the retrosynthetic perspective, we envisioned epoxide 22 as a common precursor of 19 and 20 by the disconnection via ethynylation, ring-closing metathesis, and asymmetric allylation or pentenylation. We expected that either the methyl group or the aminoethyl group on the C8-quaternary center could be installed via nucleophilic opening of the epoxide with hydride or cyanide ion. To access 22, we envisaged the approach from aldehyde 24 involving the asymmetric MBH reaction7,8) giving 23 and diastereoselective epoxidation.
Our synthesis thus began with the application of the β-ICD-HFIPA method to aldehyde 25, which afforded 26 in excellent enantiomeric purity (99% ee) in 58% yield together with the corresponding dioxanne (38% ee, 14% yield) (Chart 8). From MBH ester 26, epoxide 28, the pivotal intermediate corresponding to 22, was synthesized stereoselectively via the Horner–Emmons reaction and vanadium-catalyzed epoxidation. At this point, the synthetic route diverged toward fostriecin (17) and phoslactomycin B (18). That is, LiEt3BH reduction of 28 gave 29 quantitatively, from which the key ynone 19 was obtained through Yamamoto’s asymmetric allylation,53) ring-closing olefn metathesis, and addition of acetylene as the major transformations. Following the procedure we established previously, the total synthesis of fostriecin (17) was accomplished from 19 via Stille coupling of 31 with stannane 32.
On the other hand, the synthesis of phoslactomycin B started from DIBAL-H reduction of 28, which allowed the selective reduction of the ester group without affecting the epoxy group to give 33 in good yield. After Dess–Matin oxidation of 33, the corresponding aldehyde was then subjected to asymmetric pentenylation using the reagent prepared from (Z)-2-pentenylpotassium and (+)-(Ipc)2BOMe to give the syn-isomer with excellent diastereoselectivity (dr=97 : 3) in 83% yield. In this particular case, silver-catalyzed pentenylation using trimethoxy[(Z)-pent-2-enyl]silane under Yamamoto’s asymmetric allylation conditions53) did not proceed at all. The subsequent crucial nucleophilic opening of the epoxide was found to be low yielding under the common conditions using KCN, Et2AlCN, and trimethylsilyl cyanide (TMSCN). However, the LiCN–acetone complex54) was found to promote this reaction effectively. Further transformations involving LiAlH4 reduction followed by tert-butoxycarbonylation, acryloylation, and ring-closing olefin metathesis produced 34, which was converted into key ynones 20. The synthesis of phoslactomycin B (18) was then achieved via Stille coupling of 35 with stannane 36 in the same manner as described for the synthesis of fostriecin (17).

Glutmate receptors mediate the majority of excitatory synaptic transmission in the central nervous system and are involved in numerous important physiological mechanisms such as memory, learning, and synaptic plasticity.55) Importantly, excessive stimulation of these receptors is responsible for a variety of neurological disorders and neuronal damage from stroke.56) Therefore, glutamate receptor agonists and antagonists with entirely novel structures are in high demand to probe the relationship between glutamate receptors and brain mechanisms and neuronal diseases. In addition, such antagonistic and agonistic compounds also provide useful lead compounds for drug discovery. In this context, we focused on the synthesis of dysiherbaine (37), neodysiherbaine A (38), and kaitocephalin (39). The total syntheses of dysiherbaine (37) and kaitocephalin (39) are presented here.

Dysiherbaine (37) was isolated by Sakai et al. from the Micronesian sponge Dysidea herbacea and found to be a selective agonist of N-methyl-D-aspartate (NMDA)-type glutamate receptors.59) Structurally, this amino acid is characterized by a novel cis-fused hexahydrofuro[3,2-b]pyran ring system containing a glutamic acid appendage. When we started this project, the only relative configuration had been determined by detailed NMR analysis.59) Its low availability from natural sources, combined with the entirely novel molecular architecture and potent neuroexcitatory activity, spurred much research on the synthesis of dysiherbaine (37) and its congener neodysiherbaine (38) as well as their analogues.60) We envisaged tetra-substituted pyran 40 as a precursor of 37 and postulated that this intermediate could be accessed via the palladium-catalyzed Negishi coupling reaction of organozinc reagent 41 and enol triflate 42, accessible from tricyclic lactone 43 based on Jackson’s protocol61) (Chart 10). This coupling process was thought to be particularly challenging since Jackson’s methodology had not been successfully applied to highly functionalized enol triflates such as 42 at that time. In this synthetic plan, another key issue that had to be addressed was the efficient enantioselective construction of tricyclic lactone 43 with four contiguous cis stereogenic centers.

Our basic idea to access compound 43 began with asymmetric epoxidation62,63) of σ-symmetrical dialkenylcarbinol 47 and then the sequence proceeded through Mitsunobu inversion, introduction of a nitrogen atom by taking advantage of the epoxy alcohol functionality, construction of a Z-unsaturated ester, and formation of both lactone and tetrahydropyran rings. Following this synthetic strategy, we achieved the first asymmetric total synthesis of (−)-dysiherbaine (37), thereby determining the absolute structure as illustrated in Chart 11.57)

In the first-generation synthesis described above, we could not control the stereochemistry of the C4 quaternary center due to the less stereoselective formation of epoxide 50. We overcame this problem in our second-generation synthesis58) in which the cis-fused hexahydrofuro[3,2-b]pyran ring system was assembled in a completely stereocontrolled manner by the combination of Donohoe et al.’s tethered aminohydroxylation64) and our methodology involving Negishi coupling and Katsuki–Sharpless asymmetric epoxidation, followed by 5-exo-tet cyclization (Chart 12). We also achieved the total synthesis of neodysiherbaine A (38)65) based on fundamentally the same methodology as that employed for the second-generation synthesis of dysiherbaine (37).

Kaitocephalin (39) was isolated from Eupenicillium shearii PF1191 by Seto and colleagues68) as the first naturally occurring glutamate receptor antagonist. Due to the potent antagonistic activity against AMPA and NMDA glutamate receptors, this compound is expected to be a promising lead compound for developing therapeutic agents against various neuronal diseases such as epilepsy, Parkinson’s disease, and Alzheimer’s disease.56) However, detailed neurobiological studies and structure–activity relationship studies remain hampered because the fungus has not produced a sufficient amount of kaitocephalin (39). Under this situation, there have been a number of synthetic studies including total syntheses achieved by several groups including ours.67,69–75)
We envisaged 58 as a precursor of 39 by focusing on three carboxylic acids that could be available by oxidative cleavage of a phenyl group and a cyclopentene ring simultaneously. We then postulated that this intermediate could be accessed from 59 via rhodium-catalyzed benzylic and allylic C–H amination reactions, followed by an intramolecular nucleophilic attack of a nitrogen atom on a sulfamate group, based on Du Bois’ protocol.76,77) Since, to our knowledge, such an approach to heterocyclic compounds had not been reported previously, we became interested in probing the scope and limitation of this particular pyrrolidine synthesis.78)

We prepared a panel of various substitution patterns of cyclic sulfamates 61 from alcohols 60 by sulfamation, rhodium-catalyzed C–H amination, and butoxycarbonylation and explored the feasibility of their cyclizations. As a result, we determined the optimum conditions shown in Chart 14. Thus, upon treatment of 61 with NaH followed by water, SN2-type cyclization instantaneously took place to give cyclic compounds 62 in good to excellent yields. It is important to note that when only NaH was employed for the cyclization of 61, no 62 was produced. Although the role of water is not clear, hydrogen bonding interactions are possibly one of the main factors that influence the reactivity of the process. This method was found to be applicable to the synthesis of a variety of substituted pyrrolidines as well as piperidine, tetrahydrofuran, and tetrahydrothiophen derivatives.

Based on the pyrrolidine synthesis described above, we achieved the total synthesis of kaitocephaline (39)67) (Chart 15). Thus, sulfamate 66, prepared from the known iodoenone 63 via Suzuki–Miyaura coupling with borane 64, followed by Overman rearrangement, was subjected to our pyrrolidine synthesis involving rhodium-catalyzed C–H amination, Boc activation, and treatment with NaH–H2O to afford pyrrolidine 67 in good overall yield. Compound 67 was then converted to the advanced key intermediate 69 via another rhodium-catalyzed C–H amination reaction of 68 for the introduction of a nitrogen atom at the allylic position of the cyclopentene ring. Finally, kaitocephalin (39) was synthesized from 69 in a six-step sequence involving the formation of 3,5-dichloro-4-hydroxybenzamide and oxidative cleavage of the phenyl group and the cyclopentene ring, generating three carboxylic acids.

The importance of nitrogen-containing heterocycles as drugs and other various chemical entities continues to inspire the development of tactical methods for their synthesis. In connection with a project81–84) directed toward the synthesis of biologically intriguing natural products with highly functionalized pyrrolidinone cores such as lactacystin (72), salinosporamide A (73), neooxazolomycin (74), and oxazolomycin A (75), we became interested in developing a novel approach relying upon the Conia-ene reaction of amidomalonate 76 to give 78 via 77 (Chart 16). We envisaged that the suitably functionalized pyrrolidinone skeletons useful for the synthesis of the alkaloids 72–75 would be accessible from 78 by the discrimination of the geminal esters and stereoselective functionalization of the exo olefin.

Conia-ene reactions85) usually require harsh conditions so that racemization of the product and isomerization of the exo olefin from the β,γ- to α,β-position are serious matters of concern in the reaction shown in Chart 16. Recently, in place of the original thermal Conia-ene reaction, a number of metal-catalyzed reactions that are carried out under mild conditions have been devised for the preparation of carbocycles as well as heterocycles, although the latter are largely limited to 3-methylene-pyrrolidines and tetrahydrofurans. However, it was unknown whether metal-catalyzed versions of the Conia-ene reaction would be applicable to our envisaged transformation.
We therefore first examined the reactions of amide 79a using Au(I),86) Ni(II)/Yb(III),87) and In(III)88,89) as catalysts (Chart 17). Although Au(I)- and Ni(II)/Yb(III)-catalyzed conditions did not give satisfactory results (methods A and B), In(OTf)3 was found to catalyze the cyclization effectively to give pyrrolidinone 80a in nearly quantitative yield (method C). The formation of 80b–d revealed that this In(OTf)3-catalyzed reaction was also applicable to nonterminal alkynes and racemizable chiral alkynes. It should be emphasized that the cyclization proceeds with complete E-selectivity and without serious racemization. The addition of an equimolecular amount of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) relative to In(OTf)3 markedly accelerates the reaction (method D) and mostly results in better yields. Importantly, no endo cyclization and no isomerization of the olefinic double bond from the β,γ- to α,β-position were observed in this reaction. Treatment of 79e with In(OTf)3 or In(OTf)3-DBU did not promote the cyclization, thus suggesting that a malonyl functionality is essential for this cyclization. Furthermore, this method was found to be applicable to the synthesis of other five- to seven-membered heterocycles such as piperidinone 80f, azepanone 80g, pyrrolidines 80h and 80i, piperidine 80j, tetrahydroisoquinoline 80k, and even oxygen-containing heterocycles tetrahydrofuran 80l and tetrahydropyran 80m in moderate to excellent yields. It should be stressed that even basic products 80i–k were obtained in good yields.

Chart 18 illustrates a plausible mechanism for the above-mentioned In(OTf)3-catalyzed reaction. Nakamura and co-workers89) proposed a reaction mechanism involving carbometalation of an indium enolate with an acetylenic triple bond for the related intermolecular In(OTf)3-catalyzed reaction. Following this reaction mechanism, we rationalized the observed complete E-selectivity as well as the indispensability of a malonyl functionality, i.e., a malonic ester moiety of 76 reacts with In(OTf)3 to form indium enolate 81, which in turn undergoes an intramolecular carbometalation reaction with an alkyne to furnish alkenylindium species 82. Intermediate 82 then undergoes protonation from another molecule of 76 to produce pyrrolidinone 83 with the E-configuration with regeneration of indium enolate 81.

Salinosporamide A (73),90) a secondary metabolite of the marine actinomycete Salinospora tropica, displays very potent proteasome–inhibitory activity (IC50=1.3 nM) that is 35-fold more potent than that of omuralide, a representative proteasome inhibitor. In addition, this compound shows very potent in vivo cytotoxicity against many tumor cell lines and is therefore currently in clinical trails for the treatment of cancer. Structurally, salinosporamide A (73) is related to omuralide and comprises a γ-lactam–β-lactone bicyclic ring system with cyclohexenylcarbinol, chloroethyl, and methyl substituents. This unique molecular structure results in specific and mechanistically important interactions within the proteasome active site. The combination of the fascinating biological profile and structural complexity has attracted intense interest in the chemical and biological research communities.91–97)
Our strategy for the synthesis of salinosporamide A (73) relies on racemization-free indium-catalyzed cyclization of chiral amide 84 (Chart 19). We envisaged that the C3 quaternary center could be constructed stereoselectively from cyclized compound 85 by intramolecular delivery of an oxygen atom from the C2 substituent to the exo olefin, and another C4 quaternary center would be created by selective reduction of the ester located in the convex face of the rigid bicyclic structure. Then, according to Corey’s protocol,98) the synthesis of salinosporamide A (73) would be achieved from advanced intermediate 86 via 87 by stereoselective cyclohexenylation, followed by suitable functional group interconversions.

The required amide 90, a precursor of the key In(OTf)3-catalyzed cyclization, was prepared from chiral propargyl alcohol 88 (Chart 20) by taking advantage of Marshall’s chiral transfer methodology99) which involves the C–C bond-forming reaction of an aldehyde and an allenylzinc species. Surprisingly, during purification by silica gel column chromatography using hexane/AcOEt as the eluent, amide 90 partially underwent cyclization to give an inseparable 72 : 28 mixture of 90 and 91. When this mixture was again subjected to silica gel column chromatography conditions, pyrrolidinone 91 was obtained in very high yield. The enantiomeric purity (90% ee) of 91 suggested the production by a silica gel-promoted Conia-ene reaction rather than cyclization through the corresponding achiral allenylamide. Treatment of this mixture with a catalytic amount of In(OTf)3 in boiling toluene led to complete conversion of 90 to 91 in almost quantitative yield. It is important to note that, in this particular case, no significant loss of the enantiomeric purity was observed. Since compound 91 was very labile under basic conditions, the acetoxy group was hydrolyzed under mild lipase-catalyzed conditions to give the alcohol, which was then oxidized to aldehyde 92. For the assembly of the C3 quaternary center, 92 was subjected to acetal-mediated cationic cyclization as reported by Danishefsky and Endo.100) Thus, aldehyde 92 was treated with phenylselenenyl bromide and AgBF4 in the presence of benzyl alcohol to give the selenide, which, upon radical deselenenylation, afforded cyclic acetal 93. NaBH4 reduction of 93 resulted in excellent discrimination of the C4 geminal esters, and aldehyde 94 was obtained as the sole product after Dess–Martin oxidation. Reaction of 94 with cyclohex-2-enylzinc chloride under the Corey protocol98) yielded alcohol 95 as a single stereoisomer. Finally, the synthesis of salinosporamide (73) was achieved from 95 in a six-step sequence involving the removal of the p-methoxybenzyl group, reductive opening of the cyclic acetal, (Me2AlTeMe)2-promoted dealkylative cleavage101) of the methyl ester, β-lactonization, and chlorination.

Oxazolomycin A and neooxazolomycin, originally isolated from a strain of Streptomyces by the group of Uemura in 1985,102,103) constitute a family of structurally unique oxazole polyene lactam–lactone antibiotics together with seven other congeners identified to date (Chart 21). These oxazolomycins exhibit wide-ranging, potent inhibitory activity against Gram-positive bacteria and antiviral activity against vaccinia, herpes simplex type I, and influenza A (WSN; H1N1) as well as in vivo antitumor activity that is notable when combined with their low toxicity. The characteristic β-lactone–γ-lactam motif draws much attention due to the structural similarity with the pharmacophores of omuralide and salinosporamide A, representative 20S proteasome inhibitors. Due to the intriguing biological activities and structural challenges, a number of efforts have been dedicated to the synthesis of the oxazolomycins and their analogues.104,105) However, Kende et al.’s total synthesis106) of neooxazolomycin (74) had long been the only achievement until we have recently reported the syntheses of neooxazolomycin (74)82) and oxazolomycin A (75).84) Recently, Taylor and colleagues have also reported the formal synthesis of neooxazolomycin (74).107)

To develop an effective general method for the synthesis of the oxazolomycin family of compounds, we initially selected neooxazolomycin (74) as a target. Our synthetic plan for this natural product made the obvious disconnection at the amide linkage to the left-hand segment 96 and the right-hand segment 97 (Chart 22). Since Kende et al. demonstrated an effective methodology for the synthesis of 96106) via Stille coupling, we focused on the stereoselective construction of the right-hand segment 97. From the retrosynthetic perspective, we envisioned pyrrolidinone 100 with considerably less structural complexity as a precursor of 97 through a Nozaki–Hiyama–Kishi reaction and stereoselective dihydroxylation with concomitant lactonization. To access 100 expeditiously, we considered E-selective indium-catalyzed cyclization of amide 101. This approach is particularly appealing since the three contiguous stereogenic centers including two quaternary centers could be created in a single dihydroxylation process. We assumed that, if we were able to transform the neooxazolomycin core 98 to the oxazolomycin core 102 by cleavage of the γ-lactone, methylation of the secondary alcohol, and formation of the β-lactone, then we could utilize 98 as a common intermediate in the synthesis of most members of the oxazolomycin family.

The required amide 101 was first prepared starting from the coupling of alkyne 103 and triflate 104, both readily available from (S)-hydroxy-2-methylpropanoate (Chart 23). Upon treatment of 101 with a catalytic amount of In(OTf)3 in the presence of DBU in boiling toluene, regio- and stereoselective cyclization took place cleanly to produce 100 in good yield. The reaction again occurred with complete E-selectivity and without epimerization. In the subsequent crucial dihydroxylation of 100, we gratifyingly found that commonly used OsO4–NMO conditions promoted highly α-face-selective dihydroxylation accompanied by concomitant lactonization to yield lactone 99 as the sole product. The observed high diastereoselectivity can be explained by assuming 113 as a preferred conformer, where the approach of OsO4 is restricted to the α-face, on the basis of NOE experiments and molecular mechanics calculations.108) For the union of a right-hand core and a middle fragment, compound 99 was converted to aldehyde 107 via 106. It is noteworthy that the protection of 106 as its dioxasilinane was found to be critical for the next Nozaki–Hiyama–Kishi coupling with the middle fragment 108. For example, after hydrogenolytic debenzylation of 111, Dess–Martin oxidation of the resulting primary alcohol afforded hemiacetal 112 and it did not undergo coupling with 108, possibly because the equilibration between 112 and the corresponding aldehyde scarcely existed under the reaction conditions. This result suggested the necessity of the protection of both the C3- and C16-hydroxy groups. After considerable experimentation under various conditions, the Nozaki–Hiyama–Kishi reaction of 107 with 108 was found to be achieved best109) using 4 eq. of CrCl2 and 0.2 eq. of NiCl2 in tetrahydrofuran–dimethyl sulfoxide (THF–DMSO) at room temperature to give 109 in satisfying yield. Although no diastereoselectivity was observed in this coupling, Dess–Martin oxidation followed by L-selectride reduction allowed the highly stereoselective production of 110 with the desired R-configuration. Exposure of 110 to HF-pyridine followed by acetylation of the resulting triol furnished right-hand segment 97.

The left-hand segment 96 was prepared by the method outlined in Chart 24, which featured a marked improvement of the procedure82) we previously reported. The synthesis began with a Cinchona alkaloid-catalyzed asymmetric cyclocondensation of an aldehyde and an acid chloride. Thus, according to Nelson’s protocol,110) aldehyde 114, readily available from propargyl alcohol, was reacted with propionyl chloride using 0.2 eq. of 115 and 4 eq. of LiClO4 at −78°C to give β-lactone 116 with excellent enantioselectivity (98% ee) and diastereoselectivity (>99% de). Compound 116 was then converted to 118 via 117 in a six-step sequence involving methylation, iodination, and diimide reduction as major transformations. Iodoalkene 118 was then subjected to Stille coupling with stannane 119111) using Pd(PPh3)4, CuI, and CsF in DMF at room temperature to give Z,Z,E-triene 120 in geometrically pure form. When this coupling was carried out using Pd(0) catalyst alone, some extent of isomerization of the triene system was always observed. The left-hand segment 96 was then prepared from coupling product 120 in good yield. Finally, following Kende et al.’s procedure exactly,106) condensation of 96 with the free amine in situ generated from the right-hand segment 97 followed by deacetylation allowed us to complete the total synthesis of neooxazolomycin (74).

Taking into account the labile nature of oxazolomycin A (75) under various deprotection conditions, we focused on a challenging strategy wherein the final step is the selective construction of the β-lactone ring from the unprotected tetrahydroxy acid 121 (Chart 25). Based on the methodology developed in the synthesis of neooxazolomycin (74), we envisioned the assembly of 121 from the aforementioned two parts 96 and 108 and the right-hand core 122. For aldehyde 122, we considered an approach from γ-lactam 106, the right-hand core of neooxazolomycin, via cleavage of the γ-lactone, methylation of the secondary alcohol, and appropriate protection based on the concept mentioned in the synthetic plan for neooxazolomycin (74).

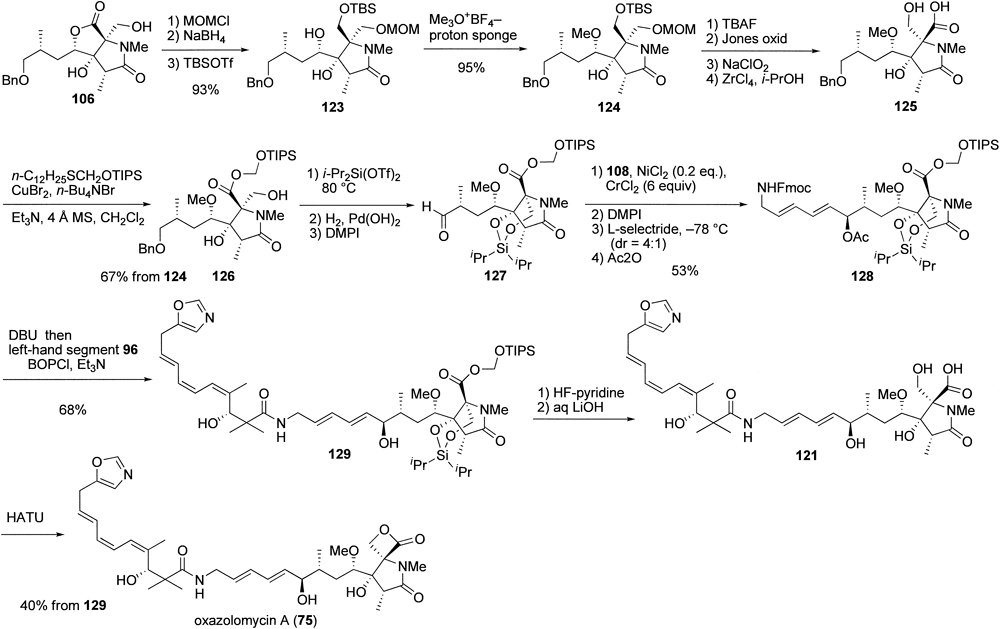
After several shorter approaches proved inefficient, the envisaged key intermediate 122 was elaborated as aldehyde 127 from 106 as illustrated in Chart 26. In this synthesis, the methylation step was critical and heavily influenced by the steric environment of the C4-hydroxy group. Eventually, we found that methylation was best achieved with 123 using Meerwein’s reagent and proton sponge to afford 124 almost quantitatively. After compound 124 was converted to hydroxy acid 125, we addressed the preparation of aldehyde 127 required for the synthesis of the right-hand segment. For the first esterification step, we chose the (triisopropylsilyl)oxymethyl (TIPSOM) ester because we expected this ester group to be easily cleaved under mild conditions.112,113) Thus, treatment of 125 with TIPSOM(dodecyl)sulfane in the presence of CuBr2, n-Bu4NBr, and triethylamine allowed selective esterification without affecting the primary hydroxy group to give ester 126 in good yield. It appeared that this esterification did not proceed selectively in the absence of triethylamine to afford the compound in which both the carboxylic acid and the primary alcohol were protected. In the same manner as described for the synthesis of neooazolomycin (74), TIPSOM ester 126 was then converted to aldehyde 127, which was further transformed into the right-hand segment 128 via Nozaki–Hiyama–Kishi coupling with dienyl iodode 108. The right-hand segment 128 was then united with the left-hand segment 96 to obtain 129. As expected, upon exposure of 129 to HF-pyridine at room temperature, the TIPSOM ester group was smoothly cleaved within 1 h to give the corresponding fully desilylated product, saponification of which afforded the desired tetrahydroxy acid 121 in nearly pure form after acidification with ion-exchange resin. Finally, 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU)-mediated lactonization of 121 allowed us to complete the first total synthesis of oxazolomycin A (75).
Our research in the area of natural product synthesis has focused on the development of new synthetic strategies and methodologies for the construction of complex molecules possessing intriguing structural and biological properties. In addition to the total syntheses presented in this review, we also achieved the syntheses of a variety of biologically active natural products with structural challenges such as febrifugine,114) viridiofungin A,115) trachyspic acid,116) cinatrin C1,117) β-erythroidine,118) NW-G01,119) inthomycins A–C,120) englerin A,121) ophiodilactones A and B,122) and marinomycin A.123) The strategies and methodologies we have developed are of great potential value in synthetic organic chemistry as well as pharmaceutical research.
I would like to express my sincere gratitude to all co-workers for their great contributions to the achievements described here. Their names are acknowledged in our publications cited in this review. This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Sciences and a Grant-in-Aid for Scientific Research on Priority Areas “Creation of Biologically Functional Molecules” and Innovative Areas “Reaction Integration” and “Advanced Molecular Transformations by Organocatalysis” from the Ministry of Education, Culture, Sports, Science and Technology of Japan.