Abstract
Two new cytotoxic dilactones, bisisorhizopodin (1) and isorhizopodin (2), together with known divalent actin depolymerizer rhizopodin (3), were isolated from the culture broth of a myxobacterium Myxococcus stipitatus. Spectroscopic analyses established that 1 and 2 are doubly and singly acyl-migrated isomers of 3, respectively, and comparison of their cytotoxicity revealed gradual decrease in the activity as the size of the ring contracted. Because the side chains of macrolide toxins uniformly block the contact between the actin protomers, the present result demonstrates substantial contribution of structurally diverse rings to the affinity of macrolide toxins for its target protein.
Myxobacteria are a distinctive group of bacteria1) that alternate their life form in response to the nutritional status: when eutrophic they take a motile and predatory lifestyle, but once eating up available food, they aggregate together and form fruiting bodies to withstand the food-depleted condition as dormant myxospores. Their secondary metabolites are highly unique and often accompanied by potent bioactivity, which make these organisms as one of the best resources for drug discovery from nature.2) In fact, ixabepilone, derived from epothilone B produced by Sorangium cellulosum, has been approved in 2007 for the treatment of metastatic breast cancer in the U.S.A.3)
Myxobacteria has been accepted as mesophilic soil bacteria, but attempts to discover new species from other environments identified halophilic,4–7) thermophilic,8,9) psychrophilic,10) or anaerobic lineages,11) revealing rather a broader habitat range for this group. And more importantly, two of the halophiles were found to produce unprecedented metabolites with a β-methoxyacrylate group such as haliangicins12–14) and miuraenamides.15,16)
As part of our program to discover new bioactive metabolites from underexploited bacterial resources, we have collected myxobacteria from hotspring environments and screened their fermentation products for cytotoxicity and antimicrobial activity. While most of the cytotoxic extracts contained myxothiazole A17,18) and exhibited broad antimycotic activity, three were found to selectively inhibit the growth of Saccharomyces cerevisiae. Intrigued by this unique activity, one of the hit strains coded YA1-5B-2, identified as Myxococcus stipitatus based on 16S ribosomal DNA (rDNA) sequence analysis, was chosen for further study. The active principles in its fermentation extract were pursued under the guidance of cytotoxicity against P388 murine leukemia, which resulted in the isolation of the known rhizopodin (3) and its two new congeners (Fig. 1). 3 is a fungicidal and cytotoxic C2-symmetric dilactone discovered from the same organism,19–22) and exerts potent bioactivity through depolymerization of actin.22,23) Actin is the most common cytoskeletal protein that undertakes motility function in eukaryotic cells, and small molecules that perturb its assembly-disassembly equilibrium are useful in dissecting the molecular mechanism of migration,24) vesicle and organelle movement,25) cell division,26) and establishment of cell shapes.27) To gain further information on the precise mechanism of actin depolymerization by 3, structure and activity of the new congeners were determined.
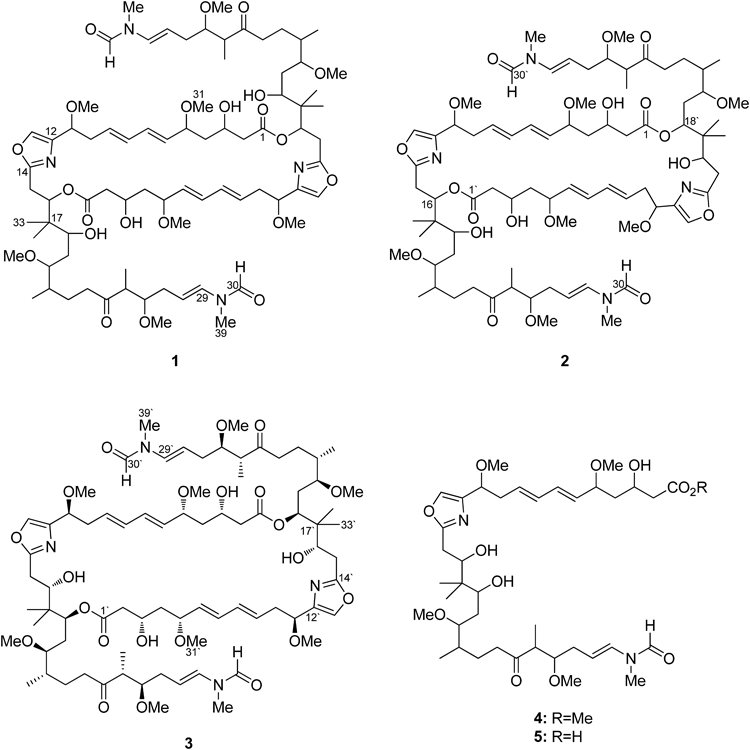
Fig. 1. Structures of Compounds 1–3
Results and Discussion
The producer strain, M. stipitatus YA1-5B-2, was isolated from sandy deposits in a sedimentation basin downstream of an outdoor hot spring bath in Toyama, Japan. It was shake-cultured in V22 medium for 8 d at 32°C, and the culture broth was extracted with n-BuOH. The concentrated extract was partitioned between 60% aqueous MeOH and dichloromethane, and the latter was further separated into 90% MeOH and n-hexane soluble fractions. The 90% MeOH layer, which exhibited potent cytotoxicity, was successively fractionated by octadecyldimethylsilyl-modified silica (ODS)-flash chromatography, gel-filtration on Sephadex LH-20, and ODS-HPLC to yield bisisorhizopodin (1; 0.4 mg), isorhizopodin (2; 0.6 mg), and the known 3 (0.3 mg).
The molecular formulae of 1–3, determined by high resolution-electrospray ionization-time-of-flight-mass spectra (HR-ESI-TOF-MS) measurements, all coincided with C78H128N4O22. In addition, their 1H-NMR spectra were quite similar with each other (Fig. 2), suggesting that the overall molecular composition is identical but difference exists in the substitution pattern. The signal duplication of an olefinic (6.72 and 7.12, H29) and formyl protons (8.30 and 8.06, H30) were again attributed to the fast cis–trans isomerization of the formamide termini, and, as is the case with 3, the abundance ratio of the cis versus trans isomers was estimated to 2 : 1 according to the integration.20)
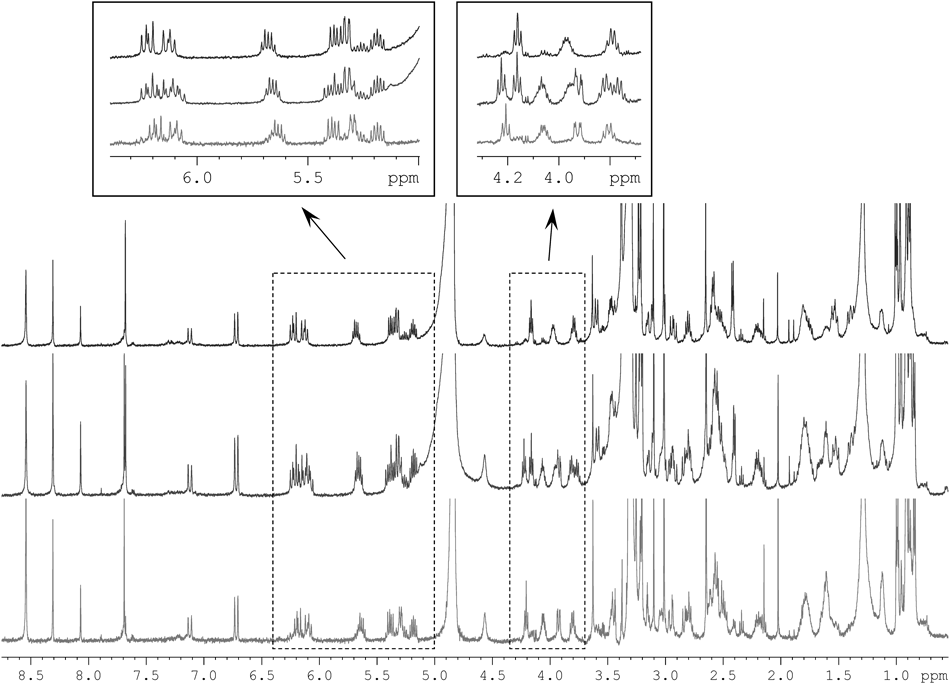
In-depth analysis of the 1H, heteronuclear-single quantum coherence (HSQC), and correlation spectroscopy (COSY) spectra of 1 revealed five fragments C2–C11, C15–C16, C18–C20, C36–(C21)–C23, and C37–(C25)–C29, which are all present in 3 (Fig. 3). Substitution of a carboxyl group at the head (C2) and an N-methyl vinylformamide at the tail (C29) of this fragment sequence, oxazole and isopropylidene interruptions, and connection between C20 and C21 were also confirmed by heteronuclear multiple-bond correlation spectroscopy (HMBC) correlations relevant to these units, retracing the same monomeric polyketide–peptide chain in 1. However, remarkable shiftings (0.1 ppm or more) of proton resonances were observed in the head (H3–H4) and middle (H15–H19) regions along this backbone. Especially, the acyloxy methine proton (H18) was upfield-shifted from 5.32 to 3.60 ppm while the proximate carbinol proton (H16) downfield-shifted from 3.92 to 5.32 ppm, suggesting migration of both the lactone linkages from C16 to C18. Although a decisive evidence was not obtained from 1 due to its scarcity, the other congener 2 endorsed this assignment by exhibiting an HMBC correlation across the lactone linkage in the same partial structure (vide infra). Thus, the structure of 1 was concluded to be a symmetrical lactone bridge-migrated isomer of 3, and was named as bisisorhizopodin following the precedent designation.28,29)

Fig. 3. Structural Fragments (Bold Lines) Deduced by COSY and Key HMBC Correlations (Arrows) Supporting the Structures of 1 and 2
In contrast, the 1H-NMR spectrum of 2 contained all signals from 1 and 3, appeared as if it were a mixture of these congeners (Fig. 2). A careful interpretation of a full set of two dimensional (2D)-NMR spectra allowed establishing both of the monomer chains in 1 and 3, and they were intercorrelated by two HMBC crosspeaks from H16 (5.32) to C1′ (173.3) and from H18′ (5.29) to C1 (171.8) (Fig. 3). Therefore, the structure of 2 proved to be a single acyl migration analog of 3.
To further ensure the structure assignments made as above, a chromatographic comparison of deacylation products from 1–3 was conducted. Compounds 1–3 (each 35 µg) were treated with 0.5 N NaOMe (35 µL) and the reaction products were subjected to LC-MS analysis. Neither of the analytes contained the expected methyl ester monomer 4: instead, an intense negative ion peak at m/z 751 was commonly detected at tR 20.8 min, supporting the production of carboxylate monomer 5 from 1–3. The precedence of saponification over transesterification is attributed to the generation of sodium hydroxide during the micro-scale reaction. Although a rigorous characterization of each product was not possible by NMR due to the scarcity of 1–3, their identity is quite reasonable in the light of polyketide biosynthesis, which is stereochemically controlled.
The occurrence of desymmetrized isomers similar to 2 was reported for sponge-derived actin-depolymerizering dilactones swinholide A28,30) and bistheonellide A.29) Because the acidic treatment of swinholide A produced singly and doubly lactone bridge-transposed derivatives, potential production of these isomers during separation on acidic silica gel was proposed.28) In this work, all purification steps were made under neutral conditions and the yields of the new congeners were higher than the parent compounds, which shows a clear contrast to the abundance of isoswinholide A and isobistheonellide A (less than 1% of the major metabolites).
The cytotoxicity of compounds 1–3 was evaluated as 50% growth inhibition (GI50) 27.2±8.1, 17.0±4.6, and 5.2±2.1 nM, respectively, showing decrease in the activity as the macrolactone ring contracted in size.
Compound 3 incorporates multiple traits that are found in actin depolymerizing cytotoxins31): oxazole rings from the tris-oxazole class toxins, N-methyl vinylformamide tails from the tris-oxazoles and the aplyronin class, and dimeric constitution from the swinholide/bistheonellide class and luminaolide.32) In fact, a crystal of actin/3 complex showed binding with two equimolar of G-actin (actin monomer) with virtually an identical mode of interaction with other related toxins at the vinylformamide tails.22)
In contrast to the clear-cut similarity in the role of the tails that physically disrupt the contact between actin monomers, ring portions of the macrolides are structurally diverse and occupy different sites on the protein surface.22,31) Nevertheless, their roles seem to be identical, because both tris-oxazoles and dimeric macrolides competed with two actin-capping proteins of the gelsolin superfamily in competition-binding experiments.33–35) Examination of the actin-bound crystals corroborated superposition of their binding interfaces, and taking results of kinetic analyses into consideration,33) a gelsolin-mimetic mode of binding was proposed for these toxins. More specifically, the ring portions work as adapters to attach to the exposed site of binding in actin filaments. This facilitates proper positioning of the tail appendages in the groove separating domain 1 and 3 in the protomers, resulting in the disassembly of the filaments.
It is very likely that 3 has the same mode of action, and decrease in the activity by simple ring contraction should reflect partial malfunction of the dilactone ring as a molecular adaptor. This is consistent with the observation that a synthetically prepared side chain of aplyronine A lacks cytotoxicity and has decreased actin-depolymerization activity.36)
Our results support the proposed function of the ring moieties of macrolide toxins in their actin depolymerization mechanism, and again illuminate sophistication of the structure of natural products, which are fine-tuned to fit in their molecular targets.
Table 1. Comparison of the NMR Spectroscopic Data (500 MHz, CD3OD) for Rhizopodins
1–3| Bisisorhizopodin (1) | Isorhizopodin (2) | Rhizopodin (3) |
|---|
| Position | δC | δH mult. (J in Hz) | HMBC | δC | δH mult. (J in Hz) | HMBC | Position | δC | δH mult. (J in Hz) | HMBC | δC | δH mult. (J in Hz) | HMBC |
|---|
| 1 | 172.3 | | | 171.8 | | | 1′ | 173.3 | | | 172.2 | | |
| 2 | 43.1 | 2.41 d (6.6) | 1, 3, 4 | 43.1 | 2.42 m | 1, 3, 4 | 2′ | 43.8 | 2.55 m | 1′, 3′ | 43.6 | 2.40 m | 1′ |
| 3 | 66.5 | 3.97 m | | 66.0 | 3.95 m | | 3′ | 66.7 | 4.06 m | | 66.7 | 4.06 m | |
| 4 | 43.3 | 1.74 m | | 43.3 | 1.75 m | 3, 5 | 4′ | 43.4 | 1.82 m | 3′, 5′ | 43.3 | 1.79 m | |
| | 1.52 m | | | 1.52 m | | | | 1.66 m | | | 1.65 m | |
| 5 | 81.0 | 3.79 m | | 80.9 | 3.79 m | 31 | 5′ | 81.0 | 3.82 m | 7′, 31′ | 81.0 | 3.80 m | |
| 6 | 131.8 | 5.37 dd (15.0, 8.3) | 8 | 132.0 | 5.37 dd (6.8, 15.0) | 8 | 6′ | 132.0 | 5.40 dd (8.1, 15.1) | 8′ | 132.2 | 5.38 dd (14.8, 8.1) | 8′ |
| 7 | 134.9 | 6.22 dd (15.0, 10.5) | 5, 6 | 134.7 | 6.22 dd (10.2, 15.1) | 5, 9 | 7′ | 134.9 | 6.18 m | 5′ | 134.6 | 6.18 dd (15.1, 10.3) | |
| 8 | 133.0 | 6.12 dd (15.0, 10.6) | | 132.9 | 6.12 dd (10.2, 15.8) | | 8′ | 133.1 | 6.08 dd (10.3, 15.6) | | 132.9 | 6.08 dd (15.1, 10.3) | |
| 9 | 131.2 | 5.68 dt (14.9, 7.3) | 7 | 131.1 | 5.66 m | 7, 11 | 9′ | 131.1 | 5.66 m | 7′, 11′ | 130.8 | 5.63 dt (15.0, 7.3) | 7′ |
| 10 | 38.5 | 2.55 m | 8, 11, 12 | 38.5 | 2.55 m | 8 | 10′ | 38.2 | 2.62 m | 8′, 11′, 12′ | 38.1 | 2.61 m | 11′ |
| 11 | 76.2 | 4.16 t (6.4) | 13, 32 | 76.4 | 4.16 t (6.6) | 9, 13, 32 | 11′ | 76.3 | 4.22 t (6.6) | 13′, 32′ | 76.4 | 4.20 t (6.5) | |
| 12 | 140.8 | | | 141.0 | | | 12′ | 139.9 | | | n.o.a) | | |
| 13 | 138.0 | 7.68 s | 12, 14 | 137.9 | 7.67 s | 12, 14 | 13′ | 137.7 | 7.68 s | 14′ | 137.5 | 7.69 s | 14′ |
| 14 | 164.1 | | | 164.2 | | | 14′ | 166.2 | | | 166.1 | | |
| 15 | 29.5 | 3.14 dd (15.4, 2.1) | 14 | 29.5 | 3.13 m | 14, 16 | 15′ | 31.7 | 2.96 m | 14′ | 31.8 | 2.94 dd (15.1, 2.6) | 14′, 16′ |
| | 2.94 dd (14.8, 10.7) | | | 2.92 m | 14 | | | 2.83 m | 14′, 16′ | | 2.82 m | |
| 16 | 76.2 | 5.32 dd (10.7, 2.0) | | 76.6 | 5.32 m | 14, 34, 1′ | 16′ | 74.0 | 3.92 dd (10.3, 2.5) | | 74.0 | 3.92 dd (10.4, 2.7) | |
| 17 | 42.1 | | | 42.0 | | | 17′ | 42.2 | | | 42.4 | | |
| 18 | 72.5 | 3.60 m | | 72.4 | 3.59 m | 33 | 18′ | 76.2 | 5.29 m | 1′ | 76.6 | 5.32 dd (8.9, 2.2) | |
| 19 | 33.2 | 1.51 m | | 33.2 | 1.51 m | 20 | 19′ | 31.6 | 1.61 m | | 31.9 | 1.60 m | |
| | 1.39 m | | | 1.39 m | | | | 1.39 m | | | | |
| 20 | 83.3 | 3.33 m | | 83.3 | 3.33 m | | 20′ | 82.7 | 3.03 m | | 82.6 | 3.03 m | 35′ |
| 21 | 35.5 | 1.80 m | | 35.5 | 1.80 m | | 21′ | 35.1 | 1.78 m | | 35.1 | 1.76 m | |
| 22 | 26.0 | 1.77 m | | 25.8 | 1.80 m | | 22′ | 25.7 | 1.78 m | | 25.6 | 1.76 m | |
| | 1.27 m | | | 1.29 m | | | | 1.26 m | | | 1.25 m | |
| 23 | 41.6 | 2.57 m | 21, 22, 24 | 41.6 | 2.58 m | 22, 24 | 23′ | 41.6 | 2.58 m | 21′, 22′, 24′ | 41.5 | 2.56 m | 22′ |
| 24 | 215.9 | | | 215.9 | | | 24′ | 215.9 | | | 215.9 | | |
| 25 | 50.0 | 2.80 m | 37 | 49.8 | 2.79 m | 24, 26, 27, 37 | 25′ | 49.8 | 2.79 m | 24, 26, 27, 37 | 49.9 | 2.79 m | 26′, 37′ |
| 26 | 83.5 | 3.46 m | 25, 37, 38 | 83.7 | 3.46 m | | 26′ | 83.7 | 3.46 m | | 83.5 | 3.46 m | 24′ |
| 27 | 30.9 | 2.50 m | 29 | 30.9 | 2.50 m | | 27′ | 30.9 | 2.50 m | | 30.9 | 2.50 m | |
| | 2.20 m | 28 | | 2.19 m | 26, 28 | | | 2.19 m | 26, 28 | | 2.20 m | |
| 28 | 107.0 | 5.18 m | | 106.8 | 5.18 m | 29 | 28′ | 106.8 | 5.18 m | 29 | 107.0 | 5.18 m | |
| 28Zb) | 109.0 | 5.26 m | | 109.1 | 5.25 m | | 28′Z | 109.1 | 5.25 m | | 109.1 | 5.26 m | |
| 29 | 131.7 | 6.72 d (13.8) | 30, 39 | 131.6 | 6,71 d (13.9) | 27, 30, 39 | 29′ | 131.6 | 6,71 d (13.9) | 27, 30, 39 | 132.0 | 6.72 d (13.9) | 30′, 39′ |
| 29Z | 126.9 | 7.12 d (14.5) | | 126.9 | 7.12 d (14.6) | 27, 39Z | 29′Z | 126.9 | 7.12 d (14.6) | 27, 39Z | 126.7 | 7.12 d (14.4) | |
| 30 | 164.2 | 8.30 s | 29, 39 | 164.3 | 8.31 s | 29, 39 | 30′ | 164.3 | 8.31 s | 29, 39 | 164.3 | 8.30 s | 29′, 39′ |
| 30Z | 162.8 | 8.06 s | | 163.0 | 8.07 s | 29Z | 30′Z | 163.0 | 8.07 s | 29Z | 163.1 | 8.06 s | |
| 31 | 55.9 | 3.21 s | 5 | 55.8 | 3.22 s | 5 | 31′ | 56.0 | 3.20 s | 5′ | 55.9 | 3.21 s | 5′ |
| 32 | 57.1 | 3.23 s | 11 | 56.7 | 3.23 s | 11 | 32′ | 56.4 | 3.26 s | 11′ | 56.3 | 3.26 s | 11′ |
| 33 | 18.7 | 0.90 s | 16, 17, 18, 34 | 18.5 | 0.91 s | 16, 17, 18, 34 | 33′ | 18.5 | 0.917 s | 16′, 17′, 18′, 34′ | 18.7 | 0.92 s | 16′, 17′, 18′, 34′ |
| 34 | 18.9 | 0.96 s | 16, 17, 18, 33 | 19.1 | 0.96 s | 16, 17, 18, 33 | 34′ | 18.5 | 0.914 s | 16′, 17′, 18′, 33′ | 18.9 | 0.92 s | 16′, 17′, 18′, 33′ |
| 35 | 58.2 | 3.38 s | 20 | 58.2 | 3.38 s | 20 | 35′ | 58.0 | 3.33 s | 20′ | 57.8 | 3.32 s | 20′ |
| 36 | 18.9 | 0.87 d (6.7) | 20, 21, 22 | 17.7 | 0.88 d (6.9) | 20, 21, 22 | 36′ | 15.6 | 0.84 d (6.9) | 20′, 21′, 22′ | 15.6 | 0.84 d (6.9) | 20′, 21′, 22′ |
| 37 | 12.6 | 0.997 d (7.0) | 24, 25, 26 | 12.6 | 0.995 d (7.0) | 24, 25, 26 | 37′ | 12.6 | 0.995 d (7.0) | 24, 25, 26 | 12.6 | 0.986 d (6.9) | 24′, 25′, 26′ |
| 37Z | 12.9 | 0.991 d (6.9) | | 12.6 | 0.988 d (7.0) | | 37′Z | 12.6 | 0.988 d (7.0) | | 12.6 | 0.992 d (7.1) | |
| 38 | 57.5 | 3.30 s | 26 | 57.5 | 3.30 s | | 38′ | 57.5 | 3.30 s | | 57.5 | 3.30 s | 26′ |
| 39 | 27.2 | 3.01 s | 29, 30 | 27.3 | 3.01 s | 29, 30 | 39′ | 27.3 | 3.01 s | 29, 30 | 27.2 | 3.01 s | 29′, 30′ |
| 39Z | 33.1 | 3.10 s | | 33.2 | 3.10 s | 29Z, 30Z | 39′Z | 33.2 | 3.10 s | 29Z, 30Z | 33.1 | 3.10 s | |
a) Not observed. b) For Z isomers at the N-methyl vinylformamide termini.
Experimental
General ProceduresUV, IR, and NMR spectra were recorded on a Hitachi U-3210, a Perkin-Elmer Spectrum 100 FT-IR spectrometer, and a 500 MHz Bruker AVANCE II instrument, respectively. HR-ESI-TOF-MS and LC-MS analyses were conducted with an Agilent 1200 HPLC-DAD system coupled to a Bruker micrOTOF mass spectrometer. The optical rotations were measured on a JASCO P-1030 polarimeter.
MicroorganismMyxococcus stipitatus strain YA1-5B-2 was isolated from sandy deposits in a sedimentation basin from an outdoor hot spring bath in Ogawa-onsen Hotspring (36°52′39.77″N, 137°38′02.28″E), Toyama, Japan, on April, 2011. A spatulaful of the specimen was placed on a sterilized filterpaper layered on medium D solidified with agar. The plate was incubated for several weeks at room temperature (ca. 25°C) until fruiting bodies of myxobacteria appeared on the surface of the inoculum or filterpaper. The upper part of a fruiting body was picked by a flame-sterilized disposable syringe needle transferred on Bennetts-2 (Bn-2) agar, and repeatedly transferred until pure. The isolate, coded YA1-5B-2, was subjected to phylogenetic analysis based on 16S rRNA gene sequence by the method described peviously.37) For a 1425 bp nucleotide sequence thus amplified was searched the closest recognized relative in the DDBJ database using BLAST algorithm, which retrieved Myxococcus stipitatus (GenBank accession No. AJ233922) with 99% identity.
FermentationThe producer strain grown on Bn-2 agar was seeded with agar substratum in V-22 liquid medium (100 mL in a K-flask) using a 10 µL disposable inoculation loop. The 30 flasks were cultured on a rotary shaker operated at 200 rpm at 30°C for 8 d.
Extraction and IsolationAt the end of the fermentation period was added 1-butanol (50 mL) to each flask, and they were shaken for another 1 h to extract metabolites. The resulting emulsion was centrifuged at 6000 rpm for 10 min, and the upper organic phase was collected by careful decantation and pipettings. The crude extract thus obtained (910 mg) was partitioned between dichloromethane and 60% aqueous MeOH, and the lipophilic layer was further separated into n-hexane and 90% aqueous MeOH. The aqueous MeOH layer (306 mg) was flash-chromatographed on ODS by a stepwise elution with aqueous MeCN (30%, 45%, 60%, 75%, 90%) containing 50 mM NaClO4 to give 5 fractions. The forth fraction was the second most cytotoxic and found to contain myxalamide A as the major active principle. The most toxic third fraction was gel-filtered on Sepahdex LH-20 with 60% MeCN, yielding the cytotoxicity at the top of eluates. This fraction was finally purified by reversed-phase HPLC (Cosmosil AR-II 10×250 mm, Nacalai Tesque) eluting for the first 5 min with 50% aqueous MeCN then raising the concentration of MeCN linearly to 60% over the next 40 min, yielding bisisorhizopodin (1; 0.4 mg), isorhizopodin (2; 0.6 mg), and rhizopodin (3; 0.3 mg), respectively.
Bisisorhizopodin (1): Amorphous solid; [α]D −79.0 (c=0.033, MeOH). UV λmax (MeOH) nm (log ε): 226 (5.05), 232 (5.03), 241 (4.88). IR (attenuated total reflectance (ATR)) νmax cm−1 3454, 2935, 2824, 1696, 1658, 1601, 1379, 1092, 994. HR-ESI-TOF-MS m/z: 1491.8577 [M+Na]+ (Calcd for C78H124N4NaO22: 1491.8605).
Isorhizopodin (2): Amorphous solid; [α]D −59.1 (c=0.049, MeOH). UV λmax (MeOH) nm (log ε): 229 (4.97), 231 (4.96), 240 (4.82). IR (ATR) νmax cm−1 3454, 2927, 1696, 1657, 1377, 1092, 994. HR-ESI-TOF-MS m/z: 1491.8619 [M+Na]+ (Calcd for C78H124N4NaO22: 1491.8605).
LC-MS Analysis of 1–3To each 35-µg aliquot of rhizopodins 1–3, precooled on ice in v-bottom vials, was added 50 µL of similarly ice-cooled 0.51 M sodium methoxide. The vials were stirred for 3 min at 0°C, after which 51 µL of 1 N HCl was added to quench the reaction. MeOH was removed under a stream of Ar and the resulting aqueous solutions were extracted four times with 100 µL of EtOAc. The organic extracts were combined and blown to dryness, which was dissolved in 35 µL of MeCN and subjected to LC-MS analysis. 0.3 µL aliquots of the analytes were chromatographed on a Cosmosil AR-II column (2.0×150 mm) under the elution program of 25% MeCN in 0.1% aqueous HCO2H for 5 min, 1%/min MeCN concentration ramp to 45%, and the same concentration held for 5 min at a flow rate of 0.2 mL/min. Negative ESI was employed for mass spectral detection and elution of 5 was traced by molecular ion at m/z 751.4 [M−H]−. Retention times for 5 from 1–3 were all 20.8 min.
Cytotoxicity TestingCytotoxicity of 1–3 was evaluated using a trypan blue dye-exclusion technique. P388 murine leukemia cells were maintained in RPMI1640 medium supplemented with 10%v/v fetal bovine serum, 0.1 mg/mL kanamycin sulfate, and 10 µM 2-mercaptoethanol. To each cell suspension prepared at a density of 5×104 cells/mL was added a serially diluted drug solution, and 100 µL aliquots of test cultures were triplicated in a 96-well plate. After incubating the plate at 37°C in a 5% CO2–95% air atmosphere for 2 d, a 100 µL-volume of 0.04% trypan blue in phosphate buffered saline (PBS) was added to each well, and the number of live cells (nucleus not stained) were counted under microscopic observation. The IC50 values were calculated from triplicated data points taken at each drug concentration.
Acknowledgment
This work is supported by Grant-in-Aid for Scientific Research (Grant 23710259) to N.O. from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a fund from Toyama Prefecture. We thank Asahi-machi Board of Education, Toyama, for permitting sample collection in its cultural heritage area.
1D- and 2D-NMR spectroscopic data for compounds 1 and 2. This material is available free of charge via the Internet at https://www.jstage.jst.go.jp/browse/cpb.
References
- 1) Shimkets L. J., Dworkin M., Reichenbach H., “The Prokaryotes, Third Edition,” Vol. 7, ed. by Falkow S., Rosenberg E., Schleifer K.-H., Stackebrandt E., Dworkin M., Springer, New York, 2006, pp. 31–115.
- 2) Diez J., Martinez J. P., Mestres J., Sasse F., Frank R., Meyerhans A., Microb. Cell Fact., 11, 52 (2012).
- 3) Conlin A., Fornier M., Hudis C., Kar S., Kirkpatrick P., Nat. Rev. Drug Discov., 6, 953–954 (2007).
- 4) Fudou R., Jojima Y., Iizuka T., Yamanaka S., J. Gen. Appl. Microbiol., 48, 109–116 (2002).
- 5) Iizuka T., Jojima Y., Fudou R., Hiraishi A., Ahn J.-W., Yamanaka S., Int. J. Syst. Evol. Microbiol., 53, 189–195 (2003).
- 6) Iizuka T., Jojima Y., Fudou R., Tokura M., Hiraishi A., Yamanaka S., Syst. Appl. Microbiol., 26, 189–196 (2003).
- 7) Iizuka T., Jojima Y., Hayakawa A., Fujii T., Yamanaka S., Fudou R., Int. J. Syst. Evol. Microbiol., 63, 1360–1369 (2013).
- 8) Gerth K., Müller R., Environ. Microbiol., 7, 874–880 (2005).
- 9) Iizuka T., Tokura M., Jojima Y., Hiraishi A., Yamanaka S., Fudou R., Microbes Environ., 21, 189–199 (2006).
- 10) Dawid W., Gallikowski C. A., Hirsch P., Polarforschung, 58, 271–278 (1988).
- 11) Sanford R. A., Cole J. R., Tiedje J. M., Appl. Environ. Microbiol., 68, 893–900 (2002).
- 12) Fudou R., Iizuka T., Yamanaka S., J. Antibiot., 54, 149–152 (2001).
- 13) Fudou R., Iizuka T., Sato S., Ando T., Shimba N., Yamanaka S., J. Antibiot., 54, 153–156 (2001).
- 14) Kundim B. A., Itou Y., Sakagami Y., Fudou R., Iizuka T., Yamanaka S., Ojika M., J. Antibiot., 56, 630–638 (2003).
- 15) Ojika M., Inukai Y., Kito Y., Hirata M., Iizuka T., Fudou R., Chem. Asian J., 3, 126–133 (2008).
- 16) Iizuka T., Fudou R., Jojima Y., Ogawa S., Yamanaka S., Inukai Y., Ojika M., J. Antibiot., 59, 385–391 (2006).
- 17) Gerth K., Irschik H., Reichenbach H., Trowitzsch W., J. Antibiot., 33, 1474–1479 (1980).
- 18) Trowitzsch W., Reifenstahl G., Wray V., Gerth K., J. Antibiot., 33, 1480–1490 (1980).
- 19) Sasse F., Steinmetz H., Hofle G., Reichenbach H., J. Antibiot., 46, 741–748 (1993).
- 20) Jansen R., Steinmetz H., Sasse F., Schubert W.-D., Hagelüken G., Albrecht S. C., Müller R., Tetrahedron, 49, 5796–5799 (2008).
- 21) Horstmann N., Menche D., Chem. Commun. (Camb.), 2008, 5173–5175 (2008).
- 22) Hagelüken G., Albrecht S. C., Steinmetz H., Jansen R., Heinz D. W., Kalesse M., Schubert W.-D., Angew. Chem. Int. Ed., 48, 595–598 (2009).
- 23) Gronewold T. M. A., Sasse F., Lünsdorf H., Reichenbach H., Cell Tissue Res., 295, 121–129 (1999).
- 24) Vallenius T., Open Biol., 3, 130001 (2013). http://rsob.royalsocietypublishing.org/content/3/6/130001.full cited Aug. 13, 2013.
- 25) Thomas C., Front. Plant Sci., 3, 188 (2012). http://www.frontiersin.org/Plant_Traffic_and_Transport/10.3389/fpls.2012.00188/full cited Aug. 14, 2013.
- 26) de Lanerolle P., J. Cell Sci., 125, 4945–4949 (2012).
- 27) Michael M., Yap A. S., Semin. Cell Dev. Biol., 24, 298–307 (2013).
- 28) Kobayashi M., Tanaka J., Katori T., Kitagawa I., Chem. Pharm. Bull., 38, 2960–2966 (1990).
- 29) Kobayashi J., Tsukamoto S., Tanabe A., Sasaki T., Ishibashi M., J. Chem. Soc., Perkin Trans. I, 1991, 2379–2383 (1991).
- 30) Kobayashi M., Tanaka J., Katori T., Matsuura M., Kitagawa I., Tetrahedron Lett., 30, 2963–2966 (1989).
- 31) Allingham J. S., Klenchin V. A., Rayment I., Cell. Mol. Life Sci., 63, 2119–2134 (2006).
- 32) Kitamura M., Schupp P. J., Nakano Y., Uemura D., Tetrahedron Lett., 50, 6606–6609 (2009).
- 33) Tanaka J., Yan Y., Choi J., Bai J., Klenchin V. A., Rayment I., Marriott G., Proc. Natl. Acad. Sci. U.S.A., 100, 13851–13856 (2003).
- 34) Klenchin V. A., Allingham J. S., King R., Tanaka J., Marriott G., Rayment I., Nat. Struct. Biol., 10, 1058–1063 (2003).
- 35) Klenchin V. A., King R., Tanaka J., Marriott G., Rayment I., Chem. Biol., 12, 287–291 (2005).
- 36) Kigoshi H., Suenaga K., Takagi M., Akao A., Kanematsu K., Kamei N., Okugawa Y., Yamada K., Tetrahedron, 58, 1075–1102 (2002).
- 37) Katsuta A., Adachi K., Matsuda S., Shizuri Y., Kasai H., Int. J. Syst. Evol. Microbiol., 55, 1851–1855 (2005).