Abstract
Phytochemical investigations of the tubers of Dioscorea bulbifera L. resulted in the isolation of nine norclerodane diterpenoids, including two new compounds, diosbulbins N (1) and P (3), a new naturally occurring compound, diosbulbin O (2), and six known ones, diosbulbins A–D, F and G (4–9). Their structures were established by spectroscopic and chemical methods. The absolute stereochemistry of 1 was determined by a modified Mosher’s method, and the absolute configuration of 2 was determined by a single-crystal X-ray diffraction analysis with CuKα irradiation. Compounds 1–3 were evaluated for in vitro cytotoxicity against five human cancer cell lines.
Dioscorea bulbifera L. (family Dioscoreaceae), which distributed widely in tropical and temperate regions, has been used historically as a Traditional Chinese Medicine to treat cancer and thyroid diseases for thousands of years.1) In Bangladesh, this plant is used by tribal people for treatment of leprosy and tumors.2) Flavonoids,3–6) norclerodane diterpenoids,7–14) and steroidal saponins15) have been found from the D. bulbifera species and most of them showed potent biological activities, such as antitumor, antifungal, and anti-inflammatory effects.4,7,8,16) The clerodane-type of diterpenoids have attracted considerable interests due to their variable stereochemistry and diverse biological activities.11,17,18) Recently, we have embarked on a phytochemical investigation on the chemical constituents from the tubers of Dioscorea bulbifera L. In this paper, we deal with the isolation and structure elucidation of compounds 1–3 on the basis of the spectroscopic and chemical methods. Their cytotoxic activities were also evaluated against five human cancer cell lines, including HL-60, SMMC-7721, A-549, MCF-7, and SW-480 using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method.
Results and Discussion
The 80% EtOH extract of dried crushed tubers of D. bulbifera (30 kg) was suspended in water and successively partitioned with petroleum ether, EtOAc, and n-butanol. The EtOAc soluble portion (760 g) was purified by repeated column chromatography to afford two new norclerodane diterpenoids, diosbulbins N (1) and P (3), one new naturally occurring compound, diosbulbin O (2), and six known compounds, diosbulbins A (4),12) B (5),10,12) C (6),12) D (7),13) F (8),10,13) and G (9).10,13,14) The structures of compounds 1–3 were determined by means of spectroscopic and chemical methods.
Diosbulbin N (1) was obtained as a white amorphous powder. Its molecular formula, C19H22O7, was deduced from the high resolution-electrospray ionization-mass spectra (HR-ESI-MS) peak at m/z 385.1252 [M+Na]+ (Calcd for C19H22O7Na: 385.1258). The IR spectrum showed vibration bands at 3462 cm−1. (hydroxyl group), 1766 and 1731 cm−1 (carbonyl groups), and 876 cm−1 (furan).2)
The 13C-NMR spectrum of 1 exhibited 19 carbon resonances (Table 1). Typical signals were observed due to two ester carbonyl groups (δC 176.0 (C-17), 180.6 (C-18)), a β-substituted furan ring (δC 145.0 (C-15), 141.7 (C-16), 126.6 (C-13), and 109.7 (C-14)),9) three oxygenated sp3 methines (δC 66.0 (C-2), 78.0 (C-6), 72.3 (C-12)) and one oxygenated quaternary sp3 carbon δC 75.3 (C-8). The 1H-NMR spectrum, coupled with its 1H–1H correlation spectroscopy (1H–1H COSY) and heteronuclear single quantum coherence (HSQC) spectra, also showed the characteristic signals of a β-substituted furan ring at δH 7.62 (1H, m, H-16), 7.51 (1H, m, H-15) and 6.55 (1H, m, H-14).9) Furthermore, three downfield signals were observed at δH 5.58 (1H, dd, J=12.5, 3.0 Hz, H-12), 4.63 (1H, m, H-6) and 4.19 (1H, m, H-2) corresponding to three oxygenated methines.
Table 1.
1H- and
13C-NMR Data of
1–
3 at 400 MHz (for
1H-NMR) and100 MHz (for
13C-NMR) (δ in ppm,
J in Hz)
| Position | 1a) | 2b) | 3b) |
|---|
| δC | δH | δC | δH | δC | δH |
|---|
| 1 | 30.9 (t) | 1.80 (br d, 13.4, H-1eq) | 29.4 (t) | 2.23 (overlap, H-1eq) | 35.6 (t) | 2.16 (br d, 13.6, H-1eq) |
| | | | 1.50 (m, H-1ax) | | 1.42 (m, H-1ax) |
| 2 | 66.0 (d) | 4.19 (br s) | 77.7 (d) | 4.82 (t, 5.0) | 65.2 (d) | 4.16 (br s) |
| 3 | 30.2 (t) | 2.27 (br d, 15.0, H-3eq) | 39.7 (t) | 2.36 (m, H-3eq) | 33.2 (t) | 2.10 (br d, 15.0, H-3eq) |
| | 1.88 (ddd, 15.0, 7.9, 3.7, H-3ax) | | 1.75 (m, H-3ax) | | 1.77 (ddd, 15.0, 5.9, 4.3, H-3ax) |
| 4 | 40.8 (d) | 2.90 (m) | 43.7 (d) | 2.83 (m) | 43.4 (d) | 3.04 (m) |
| 5 | 38.8 (d) | 2.53 (ddd, 12.3, 6.4, 4.4) | 43.3 (d) | 2.02 (m) | 43.1 (d) | 1.91 (m) |
| 6 | 78.0 (d) | 4.63 (ddd, 6.0, 4.4, 3.8) | 78.5 (d) | 4.79 (d, 5.5) | 78.0 (d) | 4.66 (d, 5.7) |
| 7 | 34.9 (t) | 2.83 (dd, 15.2, 3.8, H-7eq) | 42.2 (t) | 2.47 (overlap, H-7eq) | 41.0 (t) | 2.46 (overlap, H-7eq) |
| | 2.19 (dd, 15.2, 6.0, H-7ax) | | 2.41 (overlap, H-7ax) | | 2.42 (overlap, H-7ax) |
| 8 | 75.3 (s) | — | 80.9 (s) | — | 80.7 (s) | — |
| 9 | 39.4 (s) | — | 44.6 (s) | — | 44.7 (s) | — |
| 10 | 29.8 (d) | 2.10 (td, 12.3, 2.2) | 40.5 (d) | 2.23 (overlap) | 34.6 (d) | 2.24 (td, 11.8, 2.2) |
| 11 | 38.1 (t) | 2.01 (dd, 14.7, 3.0, H-11eq) | 43.9 (t) | 2.61 (d, 14.1, H-11a) | 44.2 (t) | 2.81 (d, 14.1, H-11a) |
| | 2.18 (dd, 14.7, 12.5, H-11ax) | | 3.35 (d, 14.1, H-11b) | | 3.41 (d, 14.1, H-11b) |
| 12 | 72.3 (d) | 5.58 (dd, 12.5, 3.0) | 195.3 (s) | — | 195.8 (s) | — |
| 13 | 126.6 (s) | — | 130.8 (s) | — | 130.7 (s) | — |
| 14 | 109.7 (d) | 6.55 (dd, 1.6, 0.8) | 110.1 (d) | 7.09 (dd, 1.6, 0.4) | 110.1 (d) | 7.07 (br d, 1.6) |
| 15 | 145.0 (d) | 7.51 (t, 1.6) | 144.7 (d) | 7.54 (t, 1.6) | 144.6 (d) | 7.52 (br s) |
| 16 | 141.7 (d) | 7.62 (m) | 150.2 (d) | 8.79 (br s) | 150.8 (d) | 8.77 (br s) |
| 17 | 176.0 (s) | — | 178.8 (s) | — | 178.6 (s) | — |
| 18 | 180.6 (s) | — | 176.9 (s) | — | 178.8 (s) | — |
| 19 | 17.3 (q) | 1.00 (s) | 14.8 (q) | 1.58(s) | 14.7 (q) | 1.48(s) |
| 20 | — | — | — | — | 53.4 (q) | 3.71(s) |
a) in CD3OD, b) in C5D5N.
The NMR spectroscopic data of 1 closely resembled those of diosbulbin G (9),13,14) showing typical resonances for a norclerodane diterpenoid skeleton.9,10) The main differences between 1 and 9 were the conformation of the B-ring/δ-lactone and the substitutent patterns at C-8 of these two compounds. Evidence for the significantly downfield shifted at C-8 from δC 43.5 in 9 to δC 75.3 in 1 indicated that a hydroxyl group was attached to C-8. This conclusion was further confirmed by the heteronuclear multiple bond correlation (HMBC) correlations of the proton resonances at δH 4.63 (H-6), 2.83 (H-7eq), 2.19 (H-7ax), 2.10 (H-10), 2.18 (H-11ax), 2.01 (H-11eq), and 1.00 (H-19) with C-8 (δC 75.3) (Fig. 2).
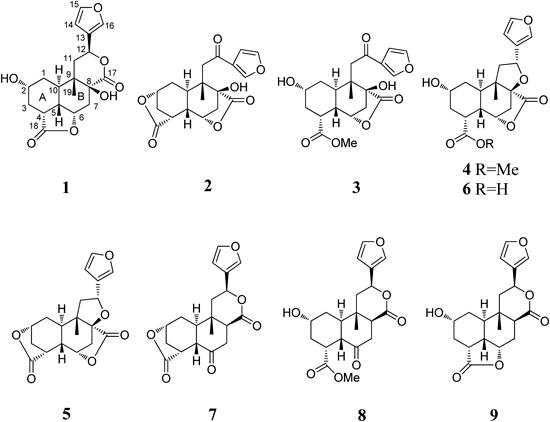
Fig. 1. Structures of Compounds 1–9

Fig. 2. Key 1H–1H COSY (Bold Bonds) and HMBC (Solid Arrows) Correlations of Compounds 1–3
The relative stereochemistry of 1 was determined on the basis of the nuclear Overhauser enhancement and exchange spectroscopy (NOESY) spectrum (Fig. 3) and by comparison to those of diosbulbin G10) and 8-epidiosbulbin G.19) The junction of the A- and B-rings was trans due to the trans-diaxial disposition of H-5 and H-10 (JH-5/H-10=12.3 Hz).19) This conclusion was further confirmed by the NOESY correlations of H-1ax, H-5 and H-7ax with Me-19, H-3ax with H-5, which indicated that both of the A/B rings were adopted as the chair-like conformations.10,19) The NOE cross peak from H-10 to H-12 indicated that the B/C (δ-lactone) rings were cis-fused.19,20) The NOESY correlations of H-4, H-6 with H-5β and their small coupling constants (JH-4/H-5=6.4 Hz, JH-5/H-6=4.4 Hz) suggested that H-4 and H-6 were both β-equatorial and the γ-lactone ring was on the α-face of the molecule.19) H-12 was in α-axial owing to a large coupling constant J=12.5 Hz with H-11ax. Thus, the relative stereochemistry of all chiral center in 1 were determined as shown in Fig. 2.
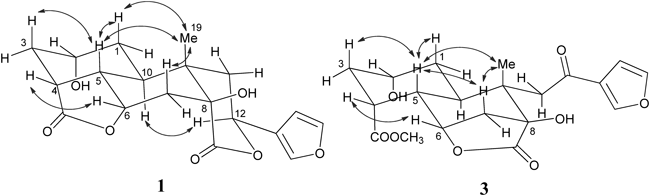
Fig. 3. Key NOESY (Arrows) Correlations of Compounds 1 and 3
Finally, the absolute configuration at C-2 of 1 was defined by a modified Mosher’s method.21) Two equal portions of 1 were derivatized with (R)-(−)- and (S)-(+)-α-methoxy-α-(trifluoromethyl)-phenylacetyl chloride (MTPA-Cl) at C-2 to yield (S)- and (R)-MTPA esters (1b and 1a), respectively. This assignment was further confirmed by the downfield shift by Δδ1b-1 1.44 and Δδ1a-1 1.33 ppm for H-2. The 1H-NMR Δδ values (Δδ=δS-MTPA-ester−δR-MTPA-ester) for the mono-MTPA esters of 1 were observed for protons adjacent to C-2, as shown in Fig. 4. According to the rule of the modified Mosher’s method, the absolute configuration of C-2 in 1 was determined to be R. Hence, the absolute configurations of the stereochemistry at C-4, C-5, C-6, C-8, C-9, C-10, and C-12 were unambigueously assigned as R, S, S, S, S, R, and S, respectively.
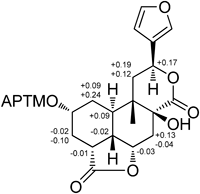
Fig. 4. Values (in ppm) of ΔδH(S–R) for the MTPA Esters of Compound 1
Diosbulbin O (2) was obtained as a colorless needles. Its molecular formula was established as C19H20O7 by HR-ESI-MS peak at m/z 383.1096 [M+Na]+ (Calcd for C19H20O7Na: 383.1101). Absorptions in the IR spectrum were attributable to a hydroxyl (3527 cm−1), a γ-lactone (1767 cm−1), a conjugated carbonyl (1663 cm−1), and a furan ring (870 cm−1). The 1H, 13C, and distortionless enhancement by polarization transfer (DEPT) NMR spectral data of 2 were similar to those of diosbulbin B (5)10,12) with characteristic signals for a β-substituted furan ring and two γ-lactones, except for the lack of tetrahydrofuran ring of 5.
The lactone carbonyl at δC 176.9 was assigned as C-18 due to the HMBC correlations with proton signals at δH 1.75 (H-3ax) and 2.02 (H-5), and the correlation with the oxymethine proton at 4.82 (H-2) indicated the lactone ring closure to C-2. Similarly the lactone carbonyl at δC 178.8 was assigned to C-17 owing to the HMBC correlations with protons at δH 2.41, 2.47 (H-7), while its 1H–1H COSY correlation with the proton at δH 4.79 (H-6) indicated the lactone ring closure to C-6. The positions of the ketone carbonyl at C-12, and the hydroxyl group at C-8 were assigned by HMBC correlations between the proton resonances at δH 2.61, 3.35 (H-11) with ketone carbonyl signal at δC 195.3 (C-12), and the proton signals at δH 4.79 (H-6), δH 2.41, 2.47 (H-7), δH 2.61, 3.35 (H-11), δH 1.58 (H-19) with the carbon at δC 80.9 (C-8), respectively.
The relative configuration of 2 was determined by the NOESY correlations. The correlation of H-1ax (δH 1.50), H-3ax (δH 1.75), H-7ax (δH 2.41), Me-19 (δH 1.58) with H-5 (δH 2.02) indicated that the A/B rings were trans-fused. Furthermore, cross-peaks from H-3ax to H-1ax, and H-5ax, from H-7ax to H-19, and H-5ax could be possible only if both of the γ-lactone rings were placed on the α face of the molecule.
In order to determine its absolute configuration, a suitable crystal of 2 was obtained from MeOH and subjected to single-crystal X-ray diffraction analysis using the anomalous scattering of CuKα radiation (CCDC 977325, Fig. 5), and the absolute configuration was established as 2R, 4R, 5S, 6S, 8S, 9S, and 10R, respectively. Although compound 2 had been reported as a synthetic derivative of diosbulbin B (5) by chemical transformation,12) herein it was isolated for the first time as a natural product. The spectroscopic data and X-ray diffraction analysis for 2 were also measured for the first time.

Fig. 5. The X-Ray Crystallographic Structure of Compound 2
Diosbulbin P (3) was obtained as a white amorphous powder. Its molecular formula was determined to be C20H24O8 on the basis of the quasi-molecular ion peak at m/z 415.1354 [M+Na]+ (Calcd for C20H24O8Na: 415.1363) in HR-ESI-MS. The IR spectrum showed absorption peaks at 3443 cm−1 (hydroxyl group), 1772 and 1664 cm−1 (carbonyl groups) and 873 cm−1 (furan). The 13C-NMR spectrum of 3 exhibited 20 signals, of which 19 were from the norclerodane diterpenoid skeleton and one from the esterified methyl group as a substitutional group. Comparison of the NMR data of 3 and 2 showed significant similarities in the chemical shifts of some carbon signals, such as C-4 (Δδ 0.3), C-5 (Δδ 0.2), C-6 (Δδ 0.5), C-7 (Δδ 1.2), C-8 (Δδ 0.2), C-9 (Δδ 0.1), C-11 (Δδ 0.3), C-12 (Δδ 0.5), and C-17 (Δδ 0.2) and the β-substituted furan ring, which suggested that the configurations of the carbons mentioned above are identical in these two compounds. Signal at δH 4.16 (H-2) in the 1H-NMR spectrum of 3 appeared in up-field by comparison with that of 2. Considering nine degrees of unsaturation, including the contribution of rings A and B, a furan ring, a γ-lactone ring, and three carbonyl groups, it suggested the cleavage of the γ-lactone ring bridged at C-2 and C-4, and the secondary hydroxyl group at C-2 and the carboxyl group at C-4 became free. The carbonyl signal at δC 178.8 was assigned as a methyl ester at C-18 due to the correlations with protons at δH 1.77 (H-3ax), δH 3.04 (H-4), δH 1.91 (H-5), and δH 3.71 (H-20) in the HMBC spectrum. The relative configuration of 3 was determined by the NOESY spectrum and by comparison of the NMR data with those of 2. The correlations of H-1ax (δH 1.42), H-3ax (δH 1.77), H-7ax (δH 2.42), Me-19 (δH 1.48) with H-5 (δH 1.91) indicated that ring A of 3 was trans-fused to ring B and both of the A/B rings were adopted as the chair-like conformations. The NOESY cross-peaks from H-7ax to H-19 and H-5ax can be established only if the γ-lactone ring bridging C-6 and C-8 was placed on the α face of the molecule. Furthermore, the NOESY correlations were depicted between the equatorial H-2 and its vicinal protons H-1 and H-3, and the lack of NOESY cross-peak of H-2 with H-10α implied that H-2 was β-equatorial. Thus, compound 3 was determined and designated as diosbulbin P.
In conclusion, two new norclerodane diterpenoids, diosbulbins N (1) and P (3), one new naturally occurring compound, diosbulbin O (2), and six known ones, diosbulbins A–D, F and G (4–9), were isolated from the tubers of Dioscorea bulbifera L. The absolute stereochemistry of 1 was determined by a modified Mosher’s method, and the absolute configuration of 2 was determined by a single-crystal X-ray diffraction analysis with CuKα irradiation. However, the absolute configuration of 3 cannot be achieved by either Mosher’s method or X-ray diffraction analysis owing to the limitation on the amount of the sample and its unstable characteristics of deterioration. The cytotoxicity assay revealed that compounds 1–3 did not show remarkable cytotoxic activities against five human cancer cell lines, HL-60, SMMC-7721, A-549, MCF-7, and SW-480 by MTT method (IC50>40 µM).
Experimental
General Experimental ProceduresOptical rotations were measured in MeOH using a Perkin-Elmer 341 polarimeter. IR spectra were recorded as KBr disks on a Bruker Vertex 70 FT-IR spectrophotometer. UV spectra were determined using Varian Cary 50. HR-ESI-MS data were measured on Thermo Fisher LC-LTQ-Orbitrap XL spectrometer. NMR spectra were recorded using a Bruker AM-400 MHz spectrometer and the 1H- and 13C-NMR chemical shifts were referred to tetramethylsilane (TMS). The X-ray diffraction experiments were carried out on a Bruker SMART APEX-II CCD diffractometer equipped with graphitemonochromatized CuKα radiation (λ=1.54178 Å). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), ODS (50 µm, YMC Co., Ltd., Japan), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden) were used for column chromatography. TLC was carried out on pre-coated silica gel GF254 plates (Yantai Chemical Industry Research Institute) and RP-C18 F254 plates (Merck, Germany). Spots were visualized under UV light (254 or 356 nm) or by spraying with 5% H2SO4 in 95% EtOH followed by heating silica gel plates or RP-C18 F254 plates. Semi-preparative HPLC was carried out on an instrument consisting of an Agilent 1100 controller, an Agilent 1100 pump, and an Agilent UV detector with a reversed-phased C18 column (5 µm, 10×250 mm, YMC-pack ODS-A). All solvents used were of analytical grade (Sinopharm Chemical Reagent Co., Ltd., China).
Plant MaterialThe tubers of D. bulbifera were collected from Hubei Province, P. R. China, in September 2011, and identified by Prof. J. Wang, College of Pharmacy, Huangzhong University of Science and Technology. A specimen (No. 20110921a) was deposited in the College of Pharmacy, Tongji Medical Center, Huangzhong University of Science and Technology.
Extraction and IsolationDried crushed tubers (30 kg) of D. bulbifera were extracted with 80% EtOH four times (v/v, 3×120 L) at room temperature. The filtrate was concentrated under vacuum to give the extract (2.2 kg), which was suspended in 10 L water and partitioned successively with petroleum ether (6×10 L), EtOAc (6×10 L), and n-BuOH (6×10 L). The EtOAc soluble portion (760 g) was subjected to silica gel column chromatography eluting with CHCl3–MeOH (50 : 1 to 0 : 1, v/v) to yield six fractions, Frs. A–F, based on TLC analysis. Diosbulbin B (5, 30 g) was obtained by recrystallization in CHCl3 from Fr. A. Fraction B was subjected to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (50 : 50, v/v), followed by chromatography over repeated silica gel column (petroleum ether–EtOAc, 50 : 50, v/v) to afford diosbulbins N (1, 30 mg) and A (4, 200 mg). Fraction C was applied to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (50 : 50, v/v), followed by chromatography on RP-C18 gel column eluted with MeOH–H2O (10 : 90 to 100 : 0, v/v), and then purified by semi-preparative HPLC (35% MeCN in H2O, flow rate 2 mL/min, wavelength 220 nm) to yield diosbulbins P (3, 1.8 mg, retention time 50 min) and G (9, 13 mg, retention time 54 min). Repeated crystallization of Fr. D yielded diosbulbin C (6, 2 g). Fractions E and F were combined and subjected to a Sephadex LH-20 column eluted with CH2Cl2–MeOH (50 : 50, v/v), followed by purification on RP-C18 gel column eluted with MeOH–H2O (10 : 90 to 100 : 0, v/v), to give diosbulbins O (2, 20 mg), D (7, 35 mg), and F (8, 15 mg).
Diosbulbin N (1): White amorphous powder; [α]D20 −12 (c=0.03, MeOH); UV λmax (MeOH) nm (log ε): 209 (3.59); IR (KBr) cm−1: 3462, 1766, 1731, 876; 1H-NMR (MeOD, 400 MHz) and 13C-NMR (MeOD, 100 MHz) see Table 1; HR-ESI-MS m/z: 385.1252 [M+Na]+ (Calcd for C19H22O7Na: 385.1258).
Diosbulbin O (2): Colorless needles; [α]D20 +74 (c=0.03, MeOH); UV λmax (MeOH) nm (log ε): 204 (4.15), 257(3.62); IR (KBr) cm−1: 3527, 1767, 1663, 870; 1H-NMR (C5D5N, 400 MHz) and 13C-NMR (C5D5N, 100 MHz) see Table 1; HR-ESI-MS m/z: 383.1096 [M+Na]+ (Calcd for C19H20O7Na: 383.1101).
Diosbulbin P (3): White amorphous powder; [α]D20 +37 (c=0.04, MeOH); UV λmax (MeOH) nm (log ε): 204 (4.00), 256 (3.42); IR (KBr) cm−1: 3443, 1772, 1664, 873; 1H-NMR (C5D5N, 400 MHz) and 13C-NMR (C5D5N, 100 MHz) see Table 1; HR-ESI-MS m/z: 415.1354 [M+Na]+ (Calcd for C20H24O8Na: 415.1363).
Preparation of the (R)- and (S)-MTPA Ester of 1Compound 1 (3.0 mg) was dissolved in 2 mL of anhydrous CH2Cl2. Dimethylaminopyridine (30 mg), triethylamine, and (S)-MTPA chloride (15 µL) were then added in sequence. The reaction mixture was stirred for 3 h at room temperature and then quenched by the addition of 1 mL of aqueous MeOH. The solution was concentrated under reduced pressure, and the residue was purified by a small silica gel column using CH2Cl2–EtOAc (75 : 25, v/v) to yield the (R)-MTPA ester of 1 (1a, 1.0 mg). The (S)-MTPA ester of 1 (1b, 1.1 mg) was prepared with (R)-MTPA chloride and purified in the same manner. The assignment was made on the basis of COSY correlations.
(R)-MTPA Ester (1a): 1H-NMR (C5D5N, 400 MHz) δ: 8.25 (1H, m, H-16), 7.64 (1H, m, H-15), 6.62 (1H, m, H-14), 5.68 (1H, dd, J=12.8, 2.7 Hz, H-12), 5.52 (1H, m, H-2), 4.81 (1H, m, H-6), 3.19 (1H, dd, J=3.7, 15.0 Hz, H-7eq), 3.08 (1H, m, H-4), 2.95 (1H, br d, J=15.0 Hz, H-3eq), 2.65 (1H, ddd, 12.3, 6.4, 4.4 Hz, H-5), 2.47 (1H, overlap, H-7ax), 2.47 (1H, overlap, H-11ax), 2.17 (1H, td, J=12.3, 1.7 Hz, H-10), 2.09 (1H, ddd, 15.0, 8.8, 4.8, H-3ax), 1.96 (1H, dd, J=14.6, 2.7 Hz, H-11eq), 1.86 (1H, br d, J=15.0 Hz, H-1eq), 1.44 (1H, m, H-1ax), 1.16 (3H, s, H-19). HR-ESI-MS m/z 601.1598 [M+Na]+ (Calcd for C29H29O9F3Na: 601.1656).
(S)-MTPA Ester (1b): 1H-NMR (C5D5N, 400 MHz) δ: 8.37 (1H, m, H-16), 7.62 (1H, m, H-15), 6.70 (1H, m, H-14), 5.85 (1H, dd, J=12.9, 2.7 Hz, H-12), 5.63 (1H, m, H-2), 4.78 (1H, m, H-6), 3.32 (1H, dd, J=3.6,15.0 Hz, H-7eq), 3.07 (1H, m, H-4), 2.85 (1H, br d, J=15.3 Hz, H-3eq), 2.66 (1H, overlap, H-11ax), 2.63 (1H, overlap, H-5), 2.43 (1H, dd, J=15.0, 5.8 Hz, H-7ax), 2.26 (1H, td, J=12.7, 2.5 Hz, H-10), 2.10 (1H, overlap, H-1eq), 2.08 (1H, overlap, H-11 eq), 2.07 (1H, overlap, H-3ax), 1.53 (1H, m, H-1ax), 1.22 (3H, s, H-19). HR-ESI-MS m/z 601.1599 [M+Na]+ (Calcd for C29H29O9F3Na: 601.1656).
X-Ray Crystallographic Analysis of 2 (CCDC 977325)The data were collected on a Bruker SMART APEX-II CCD diffractometer equipped with graphitemonochromatized CuKα radiation (λ=1.54178 Å) in the range of 3.89 to 62.49°. All non-H atoms were refined anisotropically using the full-matrix least-squares calculations on F2. Crystal was crystallized from MeOH. C19H20O7, M=360.35, 0.12×0.10×0.10 mm3, orthorhombic, P212121, T=297(2) K, a=6.13180(10) Å, b=14.4347(3) Å, c=18.4680(3) Å, α=γ=β=90°, V=1634.62(5) Å3, Z=4, Dc=1.464 Mg/m3, μ=0.942 mm−1, F(000)=760.26637 reflections collected, 2546 independent reflections (Rint=0.0394), data/restraints/parameters 2546/1/240. Goodness-of-fit on F2=1.066, R1=0.0282, wR2=0.0764 based on I>2σ(I), and R1=0.0297, wR2=0.0779 based on all data. The largest difference peak and hole were 0.107 and −0.138 eÅ−3, respectively. Absolute structure parameter: −0.06 (19).
Crystallographic data for the structure of 2 has been deposited at the Cambridge Crystallographic Data Centre (deposit number CCDC 977325). Copies of the data can be obtained free of charge on application to the CCDC, 12 Union Road, Cambridge CB2 IEZ, U.K. Fax: +44–(0)1223–336033 or e-mail: deposit@ccdc.cam.ac.uk.
Cytotoxicity AssayFive human cancer cell lines (HL-60, SMMC-7721, A-549, MCF-7, and SW-480) were used in the cytotoxic activity assay by the MTT method. Briefly, 100 µL of adherent cells (1×105) were seeded into 96-well tissue culture plates and adhere for 12 h. Each tumor cell line were treated with the medium containing different concentrations (0.0625, 0.32, 1.6, 8, 40 µM) of test compounds. After incubation with the compounds for 48 h, 15 µL of MTT solution (5 mg/mL) was added to each well and re-incubated for 4 h at 37°C. The formazan crystals produced were solubilized in dimethyl sulfoxide (DMSO). Absorption was measured at 595 nm in a 96-well microtiter plate reader (Bio-Rad 680). The experiments were carried out in triplicate (n=3), and both DDP (cis-platin, Sigma) and Taxol (Sigma) were chosen as the positive controls.
Acknowledgment
We are grateful to Dr. J. Wang at Huazhong University of Science and Technology for the authentification of this plant material. This work was financially supported by the Program for New Century Excellent Talents in University, State Education Ministry of China (NCET-2008-0224).
References
- 1) Tang Y. X., Chin. Med. J. (Engl.), 20, 435–438 (1995).
- 2) Murray R. D. H., Jorge Z. D., Khan N. H., Shahjahan M., Quaisuddin M., Phytochemistry, 23, 623–625 (1984).
- 3) Zhang J. P., Gao W., Gao H. Y., Mod. Chin. Med., 10, 34–37 (2008).
- 4) Gao H. Y., Lu Y., Wu L. J., Gao D. C., J. Shenyang Pharm. Univ., 18, 185–188 (2001).
- 5) Gao H. Y., Sui A. L., Chen Y. H., Zhang X. Y., Wu L. J., J. Shenyang Pharm. Univ., 20, 178–180 (2003).
- 6) Zhang H. M., Yuan J. Y., Herald of Medicine, 28, 490–492 (2009).
- 7) Teponno R. B., Tapondjou A. L., Ju-Jung H., Nam J. H., Tane P., Park H. J., Helv. Chim. Acta, 90, 1599–1605 (2007).
- 8) Teponno R. B., Tapondjou A. L., Abou-Mansour E., Stoeckli-Evans H., Tane P., Barboni L., Phytochemistry, 69, 2374–2379 (2008).
- 9) Wang G., Liu J. S., Lin B. B., Wang G. K., Liu J. K., Chem. Pharm. Bull., 57, 625–627 (2009).
- 10) Liu H., Chou G. X., Guo Y. L., Ji L. L., Wang J. M., Wang Z. T., Phytochemistry, 71, 1174–1180 (2010).
- 11) Shriram V., Jahagirdar S., Latha C., Kumar V., Puranik V., Rojatkar S., Dhakephalkar P. K., Shitole M. G., Int. J. Antimicrob. Agents, 32, 405–410 (2008).
- 12) Komori T., Arita M., Ida Y., Fujikura R., Kawasaki T., Sekine K., Liebigs Ann. Chem., 1973, 978–992 (1973).
- 13) Ida Y., Kubo S., Fujita M., Komori T., Kawasaki T., Liebigs Ann. Chem., 1978, 818–833 (1978).
- 14) Ida Y., Komori T., Kawasaki T., Liebigs Ann. Chem., 1978, 834–838 (1978).
- 15) Liu H., Chou G. X., Wu T., Guo Y. L., Wang S. C., Wang C. H., Wang Z. T., J. Nat. Prod., 72, 1964–1968 (2009).
- 16) Gao H., Kuroyanagi M., Wu L. J., Kawahara N., Yasuno T., Nakamura Y., Biol. Pharm. Bull., 25, 1241–1243 (2002).
- 17) Roengsumran S., Musikul K., Petsom A., Vilaivan T., Sangvanich P., Pornpakakul S., Puthong S., Chaichantipyuth C., Jaiboon N., Chaichit N., Planta Med., 68, 274–277 (2002).
- 18) Teponno R. B., Tapondjou A. L., Gatsing D., Djoukeng J. D., Abou-Mansour E., Tabacchi R., Tane P., Stoekli-Evans H., Lontsi D., Phytochemistry, 67, 1957–1963 (2006).
- 19) Rakotobe L., Mambu L., Deville A., Dubost L., Jeannoda V., Rakoto D., Bodo B., Phytochemistry, 71, 1007–1013 (2010).
- 20) Teponno R. B., Tapondjou A. L., Abou-Mansour E., Stoeckli-Evans H., Tane P., Barboni L., Phytochemistry, 69, 2374–2379 (2008).
- 21) Ohtani I., Kusumi T., Kashman Y., Kakisawa H., J. Am. Chem. Soc., 113, 4092–4096 (1991).
