Abstract
The computational structure-based drug design (SBDD) mainly aims at generating or discovering new chemical compounds with sufficiently large binding free energy. In any de novo drug design methods and virtual screening methods, drug candidates are selected by approximately evaluating the binding free energy (or the binding affinity). This approximate binding free energy, usually called “empirical score,” is critical to the success of the SBDD. The purpose of this work is to yield physical insight into the approximate evaluation method in comparison with an exact molecular dynamics (MD) simulation-based method (named MP-CAFEE), which can predict binding free energies accurately. We calculate the binding free energies for 58 selected drug candidates with MP-CAFEE. Here, the compounds are generated by OPMF, a novel fragment-based de novo drug design method, and the ligand–protein interaction energy is used as an empirical score. The results show that the correlation between the binding free energy and the interaction energy is not strong enough to clearly distinguish compounds with nM-affinity from those with µM-affinity. This implies that it is necessary to take into account the natural protein motion with explicitly surrounded by water molecules to improve the efficiency of the drug candidate selection procedure.
Because the drug discovery becomes more costly and more time-consuming despite the continuous progress of technologies, more efficient and logical drug development methods are required.1) The computational structure-based drug design (SBDD) approach2–4) is one promising prescription, and thus many methodologies and programs have been developed for the last two decades. The computational SBDD methods are categorized into two groups: the virtual screening5–8) and de novo design.9,10) The virtual screening method selects bioactive compounds from chemical compound libraries, elements of which are usually available or already known. On the other hand, the de novo drug design generates chemical compounds from scratch.
In any computational SBDD methods, drug candidates, having high binding affinity with the target protein, are selected from the chemical compound libraries or the computationally designed compound groups. For the selection, it is necessary to evaluate the binding affinity (the binding free energy) for every compound. Thus, the accuracy of the binding free energy evaluation is critical to the success of the computational SBDD. Even if the compound group includes several high-affinity compounds but if the selection method does not work well, it is impossible to discover strong candidates.
Because the standard chemical compound library includes ca. 1000000 compounds, it takes huge computational time to calculate the binding free energies for many compounds. To reduce the required computational resource, the computational SBDD usually introduce approximations to the binding free energy. (The approximate binding free energy (affinity) is often called “empirical score.”) First, the target protein is usually assumed to be rigid. This means that entropic effects, induced fit effects, and dynamical effects are totally neglected. Second, the water molecules, which often play important roles not only in forming the hydrogen bond network (water-mediated hydrogen bond)11–13) but also in the entropy loss associated with the ligand–protein docking,13–15) are not considered explicitly. The purpose of this study is to clarify how largely these approximations affect the binding free energy evaluation and to yield physical insight into the free energy evaluation, which will be helpful for interpreting the empirical score and thus selecting drug candidates properly. In addition, this will be important to improve the computational SBDD method in future.
In this paper, we examine an approximate evaluation method in comparison with an exact molecular dynamics simulation-based method, where water molecules are described with an explicit water model and the protein can fluctuate and move naturally. The accurate binding free energy evaluation methods has been extensively studied and developed based on the all-atom molecular dynamics (MD) simulations.16–19) Fujitani et al.20,21) have noticed that the Jarzynski equality leads to a reasonable foundation of the free energy calculation, and proposed a successful procedure named Massively Parallel Computation of Absolute binding Free Energy (MP-CAFEE). For several systems,20–23) the MP-CAFEE method predicted the absolute binding free energy successfully. In the case of FKBP, the root mean square (rms) error is 0.3 kcal/mol. Thus, we employed MP-CAFEE here to examine the approximations assumed by the empirical score.
Before performing the MP-CAFEE evaluation, we generated drug candidates for a typical kinase by using a newly developed fragment-based de novo design named Optimum Packing of Molecular Fragments (OPMF). Because the de novo drug design generates chemical compounds from scratch, OPMF has an advantage of being able to generate compounds with structural novelty. Interestingly, when we applied OPMF to Bcr-Abl, a tyrosine kinase leading to chronic myelogenous leukemia, we successfully reproduced its famous inhibitor, Imatinib (Gleevec). Other important features of OPMF will be discussed in the next section.
To select drug candidates from many designed molecules, the ligand–protein interaction energy is used as an empirical score. Although many empirical scores have been proposed, the interaction energy plays a central role in most of the empirical scores and therefore the results of this paper can yield physical insight into general drug candidate selection procedures. Importantly, our conclusion will be true not only for OPMF but also for other computational SBDD methods.
Experimental
OPMFTo design the drug candidates efficiently and systematically, we developed the OPMF method (Fig. 1). Because the chemical compound space is too large to survey, the OPMF method does not examine the actual fragments that consist of real elements. Instead, abstract fragments are introduced to represent homomorphous groups. For example, the charge-free planar six-membered ring is the abstract aromatic six-membered ring that represents benzene, pyridine, pyrimidine, triazine, etc. (Figs. 1a, b). In the typical case, tens of abstract fragments are used.

First, OPMF positions the abstract fragments in the protein cavity and selects the energetically stable poses (Fig. 1c). Note that all the abstract fragments are charge-free and therefore only the Lennard–Jones (LJ) interaction determines the stability at this stage. The selected fragments are connected with abstract bonds to construct abstract skeletons (Fig. 1d). Although 105 of abstract skeletons were generated initially, only 23 stable ones were selected based on the shape complementarity to the protein. The number of the skeletons would explode if one didn’t employ the abstraction of the fragment. This is because the number of abstract fragments is usually less than 50, while more than 5000 actual fragments exist. Thus, it is due to the abstraction that OPMF can conduct the systematic search in the huge chemical space.
In the next stage (Fig. 1e), real elements were assigned to the abstract skeletons to form the electrostatic interaction pair and hydrogen bond with the target protein and then the real compounds were created computationally. Here, 3759 compounds were generated. After checking the polar surface area (PSA)24) and the lipophilicity (partition function, log P)25) as well as the synthesizability, 890 compounds were selected for the target protein in this work. Note that the values of PSA and log P were calculated with Open Babel 2.3.26)
Interaction Energy Analysis as an Empirical ScoringIn order to select drug candidates, we utilized the molecular mechanics (MM) program, Tinker,27) and calculated the interaction energy between the compounds and the target protein. Here, the AMBER ff99 force field was used and the dielectric constant was set to be 4. Before calculating the interaction energy, we optimized the complex structure with the truncated Newton method,28) with the main chain of the protein constrained. Due to this procedure, the interaction energy includes the induced fit effect to some extent. First, we selected compounds interaction energies of which exceeded −40 kcal/mol. Here, 728 drug candidates were selected out.
However, the following MD calculations were too time-consuming to examine the hundreds compounds quickly. In particular, the MP-CAFEE procedure required huge computational resource even if only for one compound. To save computational time, we further discarded 558 candidates randomly with keeping the diversity of the drug candidates from the aspects of the Coulombic and vdW interaction energies and the vdW interaction energy per ligand atom. As a result, 70 compounds were selected.
Structural Stability AnalysisThe structure stability is an important feature of high affinity: If the ligand dissociates quickly from the protein in the physiological condition, we can conclude that the binding affinity is very low and don’t need the following MP-CAFEE calculations for this compound.
To check the structural stability, three 50 ns MD trajectories were conducted under the isothermal isobaric condition (T=298 K and P=1 atm), after more than 12000 TIP3P water molecules and counter ions were added to the system. Here, the Nose–Hoover thermostat and Berendsen barostat were used with the relax time constants, τT=0.5 and τP=1 ps, respectively. The system was propagated by the leap-frog method with time step of 2 fs. All the chemical bonds were constrained by the LINCS method. In order to describe the protein, the FUJI force field model,29) which is a modified version of the AMBER ff99 model based on the DF-LCCSD(T) level molecular orbital (MO) theory calculations, was employed. If RMSD values between the initial and final structures were smaller than 2.7 Å for all the three MD trajectories, we considered that the compound was stably bound to the target protein. In this work, we employed GROMACS (ver. 4.5.5)30) to perform all the MD simulations. When the ligand was stably bound, the system was quickly equilibrated, and the final structure of the MD trajectory was used for the next MP-CAFEE calculations.
By the structural stability analysis, 58 compounds were found stably bound to the target proteins out of 70 compounds. As shown in Fig. 2, the properties of the 58 compounds have reasonably wide distributions. The values of molecular weight (MW) are distributed from 240 to 350 Da, whereas the values of log P range from 1 to 4. The MW may be a little bit smaller than the standard drug, which implies that these compounds can be modified further if necessary in the future stage. The PSA, which is another property essential for Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET), was also examined. As in Fig. 2, the PSA values range from 50 to 110 Å2. All the properties satisfy the empirical rules, known as Lipinski’s rule of five31) and its extended versions,32,33) for 58 compounds, and therefore these selected compounds are likely to be suitable as drug candidates.
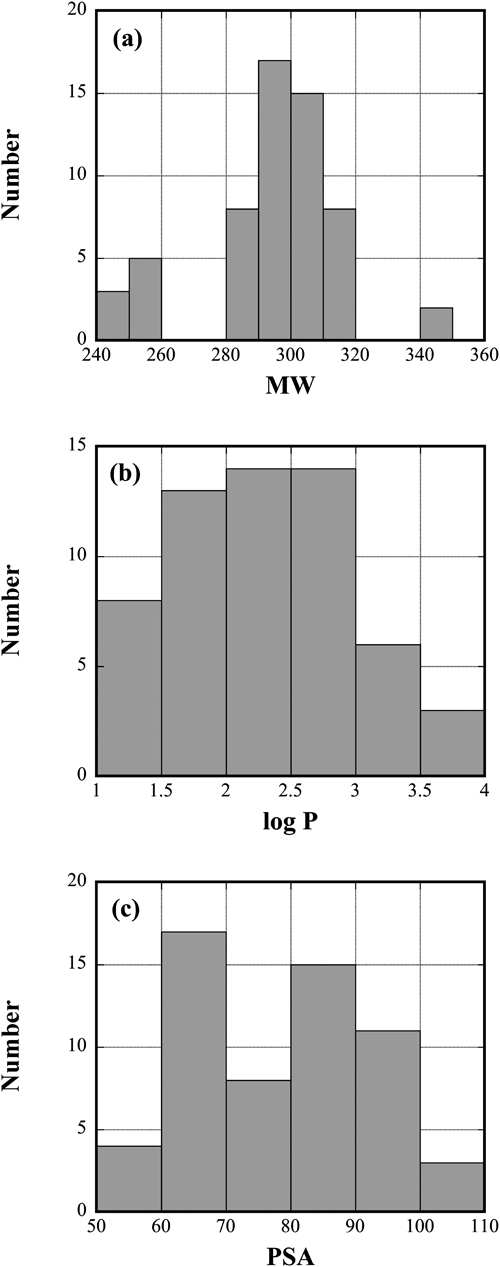
Fig. 2. Distributions of (a) MW, (b) log P, and (c) PSA
To calculate the absolute binding free energy accurately, we employed the MP-CAFEE method20,21) in this work. The MP-CAFEE method utilizes the idea of double-annihilation. The binding free energy, ΔG, corresponding to
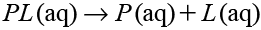 | (1) |
can be calculated as ΔG=ΔGcomp−ΔGsolv, where ΔGcomp and ΔGsolv are the free energy difference corresponding to
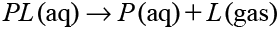 | (2) |
and
 | (3) |
respectively. Note that P, L, and PL denote the protein, ligand, and their complex, respectively, while the (aq) and (gas) mean that the molecule is in the solvation phase and in the gas phase, respectively.
The free energy changes associated with the latter two reactions can be calculated by damping the interaction between the ligand and the other. In the MP-CAFEE procedure, the scaling consists of two steps. First, the Coulombic interaction, UC, is damped by decreasing the scaling parameter, λC, from 1 to 0. In the second step, the LJ interaction, ULJ, is damped by decreasing the scaling parameter, λLJ, and finally the ligand has no interaction from the other molecules and can be considered to be in the gas phase. The scaling parameters are discretized as 11 and 21 grid points, respectively. For each grid point, 12 MD trajectories run, and therefore 384 MD trajectories are utilized for calculating one free energy difference. The free energy difference is calculated by summing up the small free energy differences between the neighboring discretized steps of the scaling parameter.
In order to obtain the free energy difference between ith state and i+1th state, we calculate the non-equilibrium work transferring from ith state to i+1th state (forward) and that from i+1th state to ith state (reverse). The Jarzynski equality, exp(−ΔG/kBT)=〈exp(−W/kBT)〉, can derive the free energy difference from the non-equilibrium work.34) This is the most important theory that justifies the MP-CAFEE procedure. Although the large statistical error might be caused by the exponential distribution, Shirts et al.35) have already solved this problem by applying the Bennett acceptance ratio (BAR) analysis, which utilizes both the forward work and the reverse work. Thus we employed the BAR to derive the free energy difference stably in the MP-CAFEE procedure. MP-CAFEE can calculate the binding free energy accurately in principle, if we equilibrate the system sufficiently in the previous step.
To calculate the binding free energy for one compound, the MP-CAFEE method requires independent 768 (=2×384) MD trajectories. Due to this feature, the MP-CAFEE method offers excellent parallelization efficiency, and therefore it is suitable for high-end computers having many CPU cores. These MD trajectories ran under the isothermal and isobaric condition (T=298 K and P=1 atm), where the Nose–Hoover thermostat (τT=1 ps) and Berendsen barostat (τP=1 ps) were used. The initial structure was taken from the 50 ns MD trajectory calculated in the previous structure stability analysis, whereas the initial velocities were generated randomly but with satisfying the Boltzmann distribution. The non-equilibrium work values were sampled in the time region from 1 to 2 ns with the interval of 100 fs. In this work, we performed the MP-CAFEE calculations for the 58 compounds selected above. Totally, this requires the 44544 (=768×58) trajectories corresponding to 89.088 µs data, and was performed on K computer (RIKEN, Japan).
Results and Discussion
Properties of the Drug CandidatesFigure 3 shows the distribution of the binding free energies calculated with the MP-CAFEE method. Several compounds exceeded µM affinity (ca. −8 kcal/mol) and two of them reached nM-affinity (ca. −12 kcal/mol). In the standard virtual screening method, only a few µM-affinity compounds were found out of thousands of compounds. Therefore, we consider that OPMF worked quite well for the present target protein. However, some other compounds have quite weak binding energies, even though they were stably bound to the target kinase through the 50 ns MD simulations and the interaction energies are stronger than −40 kcal/mol. In particular, two of them have weak binding free energies (≥−3 kcal/mol). These results indicate the importance of the selection method.

Fig. 3. Distribution of Binding Free Energy
The binding free energy may be related to many properties of compounds. For example, the hydrophobicity is considered to play an essential role in binding the hydrophobic pocket of protein. If some easily calculated property correlates to the binding free energy strongly, it will be helpful to select drug candidate efficiently. To find such properties, we examined how the binding free energy correlates to other properties (MW, PSA, and log P). As shown in Fig. 4, MW and PSA had weak correlation with the binding free energy. The correlation coefficients (denoted by R) are −0.41 and −0.43 for MW and PSA, respectively. Therefore, a compound tends to be bound to the target protein strongly, when the molecule fills the protein cavity more reasonably and the polar parts are larger. However, the correlation is not strong enough to predict the binding affinity quantitatively. Although log P is also a measure of the hydrophilicity/hydrophobicity, it doesn’t correlate to the binding free energy (R=−0.05).
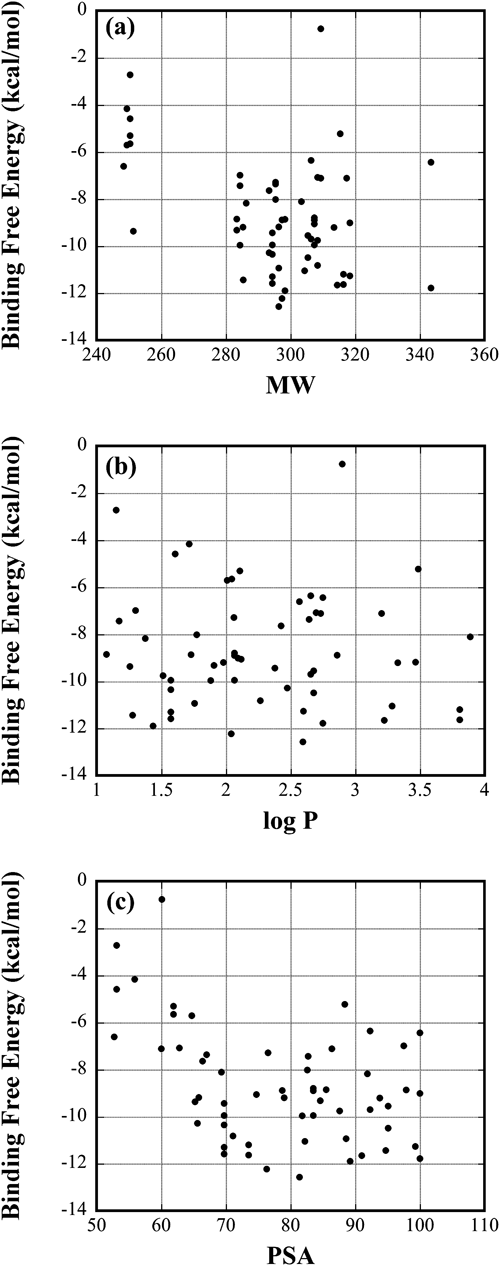
Fig. 4. Correlations of the Binding Free Energy with (a) MW, (b) log P, and (c) PSA
The binding free energy consists of the enthalpy part and the entropy part. The interaction energy is one of the most important factors of the enthalpy part, whereas the dynamic fluctuation contributes to the entropy part. Therefore, the present selection method does not consider the entropic contribution. If the ligand–protein interaction is dominant in the enthalpy part with the entropic contribution being negligible, it will be expected to correlate with the binding free energy strongly.
Figures 5a–c shows the correlation of the binding free energy to the Coulombic energy, van der Waals (vdW) energy, and the interaction energy (sum of the Coulombic energy and vdW energy) between the compound and the target protein. The Coulombic energy does not correlate to the binding free energy (R=0.05). On the other hand, the vdW interaction energy has stronger correlation with the binding free energy (R=0.38). The vdW interaction energy is dominant in the total interaction energy, and therefore the total interaction energy (sum of the Coulombic energy and vdW energy) has similar correlation (R=0.35). However, the correlation is not strong enough to clearly distinguish the nM-affinity drug candidates from others.

Fig. 5. Correlations of the Binding Free Energy with (a) Coulombic, (b) VdW, (c) Total Interaction, and (d) Distortion Energies
In addition, we found that the distortion energy of ligand weakly correlated to the binding free energy (Fig. 5d). The positive value of R (R=0.23) is attributed to the fact that the distortion energy is one term of the binding enthalpy as well as the interaction energy. If the binding pose of a compound requires larger distortion energy, the binding affinity becomes lower. Although the weak correlation suggests the importance of the other effects, it should be important to consider the distortion energy in designing drug candidates. Furthermore, the huge distortion energy sometimes indicates that the binding pose is predicted incorrectly.
The ligand–protein interaction energy of the FUJI force field (used in the MP-CAFEE calculations) is the same as that of the AMBER ff99 force field (used in the interaction energy analysis), although the FUJI force field has the modified intramolecular energy parameters. This point makes clear the meaning of the comparison. Because the interaction energy of each compound was calculated with one optimized structure and without explicitly taking into account the water molecules, the deviation between the binding free energy and the interaction energy (Fig. 5c) is dominantly attributed to the natural motion of the ligand–protein complex explicitly surrounded by the water molecules. The natural motion corresponds to the thermal equilibrium, and thus it leads to the wide energy distribution and the entropic effects.
Warren et al.7) compared the empirical scores used in the major virtual screening programs with the experimental binding affinity and showed the weak correlations between them. This result is consistent with that of the present study (Fig. 5c). However, there is a possibility that the docking pose in the reality is different from that used in the empirical score calculation, which can also cause the deviation between the empirical score and the binding affinity. In contrast, the MP-CAFEE calculation started from the same docking pose, and therefore the essential difference between the interaction energy (introduced as an approximate binding free energy) and the binding free energy is caused only by the explicit water model and the natural motion of the system. Therefore, we can conclude that the approximations to the binding free energy are fatal even if the binding pose is accurately predicted. It might be possible that the modification of the empirical parameters in the interaction energy evaluation can improve the correlation to some extent. However, we consider that the effect of the parameter modification is quite limited because the deviation is mainly caused by the different physical origins.
While drug candidates selected by the virtual screening method are usually available in the following experiments, we need to check the synthesizability of compounds generated by the de novo drug design method. The synthesizability is dependent on many factors; the stability of the compounds, the reaction rate of the designed synthetic path, the availability of raw materials, the evolution of the synthetic chemistry techniques, and even the personal knowledge, skills, and techniques of the synthetic chemist. Therefore, this remains a difficult problem of the de novo drug design. On the other hand, the MP-CAFEE method as well as other computational methods is totally free of the synthesizability problem, so we can focus only on the issue about whether or not the empirical score is a good indicator in the present study.
An additional advantage of the MD-based method is that a poor initial structure can be improved in the equilibration process to some extent, while the conventional docking methods directly suffer from the poorness and lead to unreasonable evaluation of the binding free energy. In fact, it was reported that there was a serious concern that some low quality of X-ray crystal structures can affect the docking procedure and can mislead the development and verification of the scoring function and parameters.36,37)
Concluding Remarks
The drug candidate selection step is critical to the success of the computational SBDD. In this work, we examined the empirical scoring (approximate binding free energy evaluation) in comparison with the accurate MP-CAFEE method. The advantage of the computational evaluation is free of the experimental difficulties, and thus we can make the comparison quite easily. The analyses of the MP-CAFEE calculations showed that the correlation between the ligand–protein interaction energy and the binding free energy was not strong enough to select nM-affinity drug candidates from others. This means that the accurate description of equilibrium states and the accurate evaluation of entropic contributions are necessary to predict the absolute binding free energy with the chemical accuracy. This statement will be true for other systems. In particular, quite similar results will be obtained for kinase systems, because the kinases have similar structures. To take into account these points, it is necessary to treat water molecules explicitly, because the individual water molecules can play a key role of forming the hydrogen bond network (water-mediated hydrogen bond)11–13) and the release of a water molecule from the binding pocket of protein is important for the entropic contribution in the binding free energy.13–15) To improve the drug candidate selection process, we consider that the direct application of the MD simulation-based evaluation will be promising and examine the new SBDD method currently. (A paper on this subject is under preparation.) The synthesizability and the pose selection problems still remain as important future challenges. For the latter problem, we consider that the recent MD simulation technique with high-performance computers will be a promising prescription.38)
Acknowledgment
This research has been funded by MEXT SPIRE Supercomputational Life Science (hp120297, hp130006, and hp140228) and partly by FIRST Kodama project. In particular, the huge computational resource of K computer (RIKEN, Japan) enabled us to perform all the MP-CAFEE calculations.
References
- 1) Paul S. M., Mytelka D. S., Dunwiddie C. T., Persinger C. C., Munos B. H., Lindborg S. R., Schacht A. L., Nat. Rev. Drug Discov., 9, 203–214 (2010).
- 2) Kalyaanamoorthy S., Chen Y.-P. P., Drug Discov. Today, 16, 831–839 (2011).
- 3) Anderson A. C., Chem. Biol., 10, 787–797 (2003).
- 4) Blaney J., J. Comput. Aided Mol. Des., 26, 13–14 (2012).
- 5) McInnes C., Curr. Opin. Chem. Biol., 11, 494–502 (2007).
- 6) Shoichet B. K., Nature (London), 432, 862–865 (2004).
- 7) Warren G. L., Andrews C. W., Capelli A.-M., Clarke B., LaLonde J., Lambert M. H., Lindvall M., Nevins N., Semus S. F., Senger S., Tedesco G., Wall I. D., Woolven J. M., Peishoff C. E., Head M. S., J. Med. Chem., 49, 5912–5931 (2006).
- 8) Enyedy I. J., Egan W. J., J. Comput. Aided Mol. Des., 22, 161–168 (2008).
- 9) Schneider G., Fechner U., Nat. Rev. Drug Discov., 4, 649–663 (2005).
- 10) Schneider G., Hartenfeller M., Reutlinger M., Tanrikulu Y., Proschak E., Schneider P., Trends Biotechnol., 27, 18–26 (2009).
- 11) Bairagya H. R., Mukhopadhyay B. P., Sekar K., J. Biomol. Struct. Dyn., 26, 497–507 (2009).
- 12) Nasief N. N., Tan H., Kong J., Hangauer D., J. Med. Chem., 55, 8283–8302 (2012).
- 13) Ladbury J. E., Chem. Biol., 3, 973–980 (1996).
- 14) Chiba S., Harano Y., Roth R., Kinoshita M., Sakurai M., J. Comput. Chem., 33, 550–560 (2012).
- 15) Baron R., Setny P., McCammon J. A., J. Am. Chem. Soc., 132, 12091–12097 (2010).
- 16) Khavrutskii I. V., Wallqvist A., J. Chem. Theory Comput., 7, 3001–3011 (2011).
- 17) Boyce S. E., Mobley D. L., Rocklin G. J., Graves A. P., Dill K. A., Shoichet B. K., J. Mol. Biol., 394, 747–763 (2009).
- 18) Aleksandrov A., Thompson D., Simonson T., J. Mol. Recognit., 23, 117–127 (2010).
- 19) Chodera J. D., Mobley D. L., Shirts M. R., Dixon R. W., Branson K., Pande V. S., Curr. Opin. Struct. Biol., 21, 150–160 (2011).
- 20) Fujitani H., Tanida Y., Ito M., Jayachandran G., Snow C. D., Shirts M. R., Sorin E. J., Pande V. S., J. Chem. Phys., 123, 084108 (2005).
- 21) Fujitani H., Tanida Y., Matsuura A., Phys. Rev. E Stat. Nonlin. Soft Matter Phys., 79, 021914 (2009).
- 22) Mitchell W., Matsumoto S., Curr. Opin. Chem. Biol., 15, 553–559 (2011).
- 23) Fujitani H., Shinoda K., Yamashita T., Kodama T., J. Phys.: Conf. Ser., 454, 012018 (2013).
- 24) Ertl P., Rohde B., Selzer P., J. Med. Chem., 43, 3714–3717 (2000).
- 25) Wildman S. A., Crippen G. M., J. Chem. Inf. Comput. Sci., 39, 868–873 (1999).
- 26) O’Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., Hutchison G. R., J. Cheminform., 3, 33 (2011).
- 27) Ponder J. W., TINKER, Software Tools for Molecular Design, Version 6.0 (2011).
- 28) Ponder J. W., Richards F. M., J. Comput. Chem., 8, 1016–1024 (1987).
- 29) Fujitani H., Matsuura A., Sakai S., Sato H., Tanida Y., J. Chem. Theory Comput., 5, 1155–1165 (2009).
- 30) Hess B., Kutzner C., van der Spoel D., Lindahl E., J. Chem. Theory Comput., 4, 435–447 (2008).
- 31) Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J., Adv. Drug Deliv. Rev., 23, 3–25 (1997).
- 32) Veber D. F., Johnson S. R., Cheng H. Y., Smith B. R., Ward K. W., Kopple K. D., J. Med. Chem., 45, 2615–2623 (2002).
- 33) Ghose A. K., Viswanadhan V. N., Wendoloski J. J., J. Comb. Chem., 1, 55–68 (1999).
- 34) Jarzynski C., Phys. Rev. Lett., 78, 2690–2693 (1997).
- 35) Shirts M. R., Bair E., Hooker G., Pande V. S., Phys. Rev. Lett., 91, 140601 (2003).
- 36) Warren G. L., Do T. D., Kelley B. P., Nicholls A., Warren S. D., Drug Discov. Today, 17, 1270–1281 (2012).
- 37) Davis A. M., St-Gallay S. A., Kleywegt G. J., Drug Discov. Today, 13, 831–841 (2008).
- 38) Shan Y., Kim E. T., Eastwood M. P., Dror R. O., Seeliger M. A., Shaw D. E., J. Am. Chem. Soc., 133, 9181–9183 (2011).