Regular Articles
Hepatoprotective Triterpenoid Saponins from Callicarpa nudiflora
2014 Volume 62 Issue 7 Pages 695-699

Details
2014 Volume 62 Issue 7 Pages 695-699
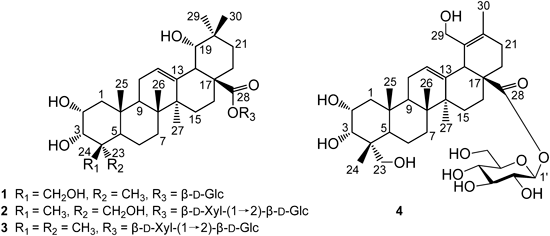
Four new triterpenoid saponins, 2α,3α,19α,24-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-glucopyranosyl ester (1), 2α,3α,19α,23-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glu-copyranosyl ester (2), 2α,3α,19α-trihydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester (3), 2α,3α,23,29-tetrahydroxyurs-12,19-dien-28-oic-acid 28-O-β-D-glucopyranosyl ester (4), together with three known compounds (5–7), were isolated from the leaves of Callicarpa nudiflora HOOK. Their structures were established by means of spectroscopic methods and chemical evidence. Hepatoprotective activities of the isolated compounds against D-galactosamine-induced toxicity have been tested. Among them, compounds 1–3 showed pronounced hepatoprotective activities against D-galactosamine-induced toxicity in WB-F344 rat hepatic epithelial stem-like cells.
Callicarpa nudiflora HOOK is distributed widely in Guangdong, Guangxi, and Hainan Provinces in China. The plant is used as traditional Chinese herbal medicines for the treatment of inflammation and bleeding.1) Previous investigation on C. nudiflora has led to the isolation of iridoids, flavonoids, triterpenoides, and phenylpropanoid glycosides.2–5) Some of them have been shown to exhibit anti-inflammatory, antibacterial, cytotoxic and hemostatic activies.6) Our previous phytochemical study of C. nudiflora yielded two triterpenoid glycosides, an acylated flavone glycoside, and three furofuran lignans.6,7) Further investigation on the 80% EtOH extract of this plant resulted in the isolation of four new compounds, 2α,3α,19α,24-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-glucopyranosyl ester (1), 2α,3α,19α,23-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester (2), 2α,3α,19α-trihydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester (3), 2α,3α,23,29-tetrahydroxyurs-12,19-dien-28-oic-acid 28-O-β-D-glucopyranosyl ester (4), along with three known compounds, luteolin (5), 5,4′-dihydroxyl-3,7,3′-trimethoxyflavone (6), caffeic acid (7). Reported herein are the isolation, structure elucidation and biological activity of these compounds.
Compound 1 was obtained as a white powder. The molecular formula, C36H58O11, was determined by high resolution-electrospray ionization-mass spectra (HR-ESI-MS) (m/z 665.3878 [M−H]−, Calcd for 665.3901). The 1H-NMR spectrum of 1 in pyridine-d5 showed six methyl singlets (δH 0.99, 1.09, 1.15, 1.19, 1.55, 1.68). Additional proton resonances observed included those ascribed to an olefinic proton at δH 5.52 (1H, br s), two oxymethine protons at δH 4.48 (1H, m) and 4.62 (1H, d, J=1.8 Hz), two oxymethylene protons at δH 3.87 and 4.15 (1H each, d, J=10.8 Hz), and an anomeric proton signal at δH 6.38 (1H d, J=8.4 Hz) correlated in the heteronuclear single quantum coherence (HSQC) spectrum with an anomeric carbons at δC 96.4 in the 13C-NMR spectrum (Table 1). The 13C-NMR spectrum of 1 displayed 36 carbon signals, of which 6 were attributed to the sugar moiety and the remaining 30 to the aglycone. Signals at δC 124.3, 144.8, and 177.7 were assigned to a pair of typical olefinic carbons and a carboxy carbonyl carbon, respectively. These spectroscopic data indicated that 1 is an oleanene-type triterpene8) with four hydroxyl groups, a trisubstituted double bond. The two oxymethylene proton signals at δH 3.87 and 4.15, which correlated with the carbon resonance at δC 65.7 in the HSQC spectrum, showed heteronuclear multiple bond connectivity (HMBC) correlations with C-3, C-4, and C-24, justifying its assignment to C-23. The assignment of hydroxyl groups at C-2 and C-3 was confirmed from the HMBC correlations from H-2 to C-4 and C-10 and from H-1, H-2, H-23, and H-24 to C-3. Comparison of the NMR spectroscopic data of 1 with sericoside9) demonstrated that two compounds were almost identical except for the A ring. The splitting pattern of H-3 (1H, d, J=1.8 Hz) suggested that 1 was the C-3 epimer of sericoside. Thus, the C-3 hydroxy group of 1 was α-orientation. From the foregoing evidences it was concluded that 1 was a glycoside of tetrahydroxyolean-12-en-28-oic acid. Acid hydrolysis of 1 with 2 M HCl afforded D-glucose, which was identified by GC analysis of its trimethylsilyl L-cysteine derivatives.10) In the HMBC spectrum of 1, the presence of correlations from Glc-H-1 (δH 6.38) to C-28 (δC 177.7) confirmed that the glucose unit is located at C-28 of the aglycone. The α-orientation of 2-OH, 3-OH, and 19-OH in 1 was deduced by analysis of the nuclear Overhauser effect spectroscopy (NOESY) spectrum which showed nuclear Overhauser effect (NOE) correlations between the following proton pairs: H-2/H-3, H-2/H-24, H-2/H-25, and H-19/H-30. Thus, compound 1 was elucidated as 2α,3α,19α,24-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-glucopyranosyl ester.
| Position | 1a) | 2b) | 3a) | 4b) | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 1.88 (1H, m) | 43.5 | 0.89 (1H, d, 12.0) | 47.8 | 1.78 (1H, t, 12.0) | 43.2 | 1.32 (1H, m) | 42.8 |
| 1.97 (1H, m) | 1.91 (1H, dd, 12.0, 4.2) | 1.92 (1H, m) | 1.67 (1H, m) | |||||
| 2 | 4.48 (1H, m) | 66.7 | 3.69 (1H, dd, 4.2, 2.4) | 69.7 | 4.34 (1H, m) | 66.6 | 3.90 (1H, m) | 67.3 |
| 3 | 4.62 (1H, d, 1.8) | 74.8 | 3.35 (1H, d, 2.4) | 78.4 | 3.77 (1H, d, 2.4) | 79.9 | 3.61 (1H, d, 2.4) | 78.8 |
| 4 | 45.6 | 44.1 | 39.4 | 44.4 | ||||
| 5 | 1.88 (1H, m) | 50.2 | 1.31 (1H, m) | 48.5 | 1.64 (1H, m) | 49.5 | 1.67 (1H, m) | 42.5 |
| 6 | 1.55 (1H, m) | 19.7 | 1.50 (1H, m) | 19.3 | 1.37 (1H, m) | 19.2 | 1.34 (1H, m) | 18.9 |
| 1.71 (1H, m) | 1.60 (1H, m) | 1.52 (1H, m) | 1.53 (1H, m) | |||||
| 7 | 1.51 (1H, m) | 34.2 | 1.30 (1H, m) | 33.4 | 1.66 (2H, m) | 33.8 | 1.34 (2H, m) | 34.5 |
| 1.62 (1H, m) | 1.63 (1H, m) | |||||||
| 8 | 41.0 | 41.0 | 40.5 | 40.8 | ||||
| 9 | 2.15 (1H, m) | 49.0 | 1.85 (1H, m) | 49.6 | 2.09 (1H, m) | 48.7 | 1.64 (1H, m) | 49.0 |
| 10 | 39.3 | 39.2 | 39.3 | 39.2 | ||||
| 11 | 2.15 (2H, m) | 25.0 | 2.0 (2H, m) | 24.9 | 2.10 (2H, m) | 24.7 | 1.81 (1H, dd, 7.8, 3.0) | 24.3 |
| 2.00 (1H, m) | ||||||||
| 12 | 5.52 (1H, br s) | 124.3 | 5.33 (1H, br s) | 124.7 | 5.51 (1H, br s) | 124.1 | 5.53 (1H, t, 3.6) | 129.2 |
| 13 | 144.8 | 144.6 | 144.9 | 138.3 | ||||
| 14 | 42.7 | 42.7 | 42.7 | 44.8 | ||||
| 15 | 1.25 (1H, m) | 29.5 | 0.92 (1H, m) | 30.1 | 1.33 (1H, m) | 29.9 | 1.14 (1H, t, 3.0) | 29.2 |
| 2.37 (1H, m) | 1.78 (1H, m) | 2.28 (1H, m) | 1.89 (1H, m) | |||||
| 16 | 2.12 (1H, m) | 28.5 | 1.76 (1H, m) | 28.0 | 2.29 (1H, m) | 28.2 | 2.00 (2H, m) | 24.6 |
| 2.82 (1H, m) | 2.28 (1H, m) | 2.81 (1H, m) | ||||||
| 17 | 47.0 | 47.2 | 47.1 | 48.2 | ||||
| 18 | 3.53 (1H, br s) | 45.1 | 3.04 (1H, br s) | 45.1 | 3.50 (1H, br s) | 45.2 | 3.25 (1H, br s) | 51.6 |
| 19 | 3.57 (1H, d, 3.6) | 81.6 | 3.27 (1H, d, 4.8) | 82.4 | 3.55 (1H, d, 3.6) | 81.7 | 133.7 | |
| 20 | 36.0 | 35.9 | 36.0 | 129.1 | ||||
| 21 | 1.04 (1H, m) | 29.5 | 1.69 (1H, m) | 29.5 | 1.03 (1H, m) | 29.6 | 2.18 (1H, m) | 25.0 |
| 2.05 (1H, m) | 1.75 (1H, m) | 2.29 (1H, m) | 2.31 (1H, m) | |||||
| 22 | 1.97 (1H, m) | 33.5 | 1.64 (1H, m) | 33.3 | 1.98 (1H, m) | 33.4 | 1.67 (1H, m) | 33.5 |
| 2.06 (1H, m) | 1.80 (1H, m) | 2.08 (1H, m) | 1.89 (1H, m) | |||||
| 23 | 1.68 (3H, s) | 24.2 | 3.29 (1H, d, 11.4) | 66.5 | 1.20 (3H, s) | 29.9 | 3.39 (1H, d, 10.2) | 71.3 |
| 3.50 (1H, d, 11.4) | 3.53 (1H, d, 10.2) | |||||||
| 24 | 3.87 (1H, d, 10.8) | 65.7 | 0.71 (3H, s) | 13.8 | 0.86 (3H, s) | 22.7 | 0.78 (3H, s) | 17.7 |
| 4.15 (1H, d, 10.8) | ||||||||
| 25 | 1.09 (3H, s) | 17.5 | 1.04 (3H, s) | 17.5 | 0.99 (3H, s) | 17.1 | 1.06 (3H, s) | 17.9 |
| 26 | 1.19 (3H, s) | 18.1 | 0.74 (3H, s) | 17.8 | 1.12 (3H, s) | 18.2 | 0.89 (3H, s) | 18.4 |
| 27 | 1.55 (3H, s) | 25.3 | 1.30 (3H, s) | 25.2 | 1.56 (3H, s) | 25.3 | 1.02 (3H, s) | 22.6 |
| 28 | 177.7 | 178.7 | 177.8 | 177.7 | ||||
| 29 | 1.15 (3H, s) | 29.2 | 0.94 (3H, s) | 28.7 | 1.13 (3H, s) | 29.2 | 3.95 (1H, d, 11.4) | 63.7 |
| 4.17 (1H, d, 11.4) | ||||||||
| 30 | 0.99 (3H, s) | 25.2 | 0.95 (3H, s) | 25.1 | 0.97 (3H, s) | 25.3 | 1.67 (3H, s) | 16.9 |
| Sugar (C-28) Glc | ||||||||
| 1 | 6.38 (1H, d, 8.4) | 96.4 | 5.45 (1H, d, 7.8) | 94.0 | 6.30 (1H, d, 8.4) | 94.2 | 5.40 (1H, d, 8.4) | 95.9 |
| 2 | 4.38 (1H, m) | 71.7 | 3.61 (1H, m) | 80.0 | 4.37 (1H, m) | 80.8 | 3.40 (1H, m) | 71.4 |
| 3 | 4.30 (1H, m) | 79.4 | 3.31 (1H, m) | 78.0 | 4.19 (1H, m) | 78.9 | 3.41 (1H, m) | 78.3 |
| 4 | 4.23 (1H, m) | 74.6 | 3.41 (1H, m) | 71.2 | 4.29 (1H, m) | 71.6 | 3.33 (1H, m) | 74.0 |
| 5 | 4.04 (1H, m) | 79.7 | 3.35 (1H, m) | 78.7 | 4.28 (1H, m) | 79.3 | 3.35 (1H, m) | 78.7 |
| 6 | 4.41 (1H, m) | 62.8 | 3.69 (1H, dd, 10.8, 4.4) | 62.3 | 4.35 (1H, m) | 62.6 | 3.68 (1H, dd, 12.0, 1.8) | 62.6 |
| 4.46 (1H, m) | 3.80 (1H, d, 10.8) | 4.40 (1H, m) | 3.81 (1H, d, 12.0) | |||||
| Xyl (1→2) Glc | ||||||||
| 1 | 4.63 (1H, d, 7.8) | 105.3 | 5.52 (1H, d, 7.2) | 106.3 | ||||
| 2 | 3.18 (1H, m) | 75.8 | 4.11 (1H, m) | 76.4 | ||||
| 3 | 3.62 (1H, m) | 78.7 | 4.34 (1H, m) | 79.5 | ||||
| 4 | 3.42 (1H, m) | 70.7 | 4.35 (1H, m) | 71.5 | ||||
| 5 | 3.15 (1H, t, 10.8) | 67.0 | 3.71 (1H, t, 10.8) | 67.8 | ||||
| 3.79 (1H, d, 10.8) | 4.39 (1H, d, 10.8) | |||||||
a) Recorded in pyridine-d5. b) Recorded in CD3OD.
Compound 2 had the molecular formula C41H66O15, as deduced from the HR-ESI-MS (m/z 821.4290 [M+Na]+, Calcd for 821.4294). The 1H-NMR spectrum of 2 was similar to those of 1 except for the configuration of C-4 and an additional set of β-D-xylose resonances. The proton signal at δH 0.71 assigned to H-24 of 2 was shifted by −0.97 ppm compared to that of 1, suggesting that the aglycone of 2 was the C-4 epimer. The hydroxymethyl at C-4 was α-oriented, as deduced from the NOESY correlation between the following proton pairs: H-2/H-24, H-2/H-25, and H-3/H-24. The linkage position of the sugar units with the aglycone was established from the following HMBC correlations: Glc-H-1 (δH 5.45)/aglycone-C-28 (δC 178.7) and Xyl-H-1 (δH 4.63)/Glc-C-2 (δC 80.0). Thus, compound 2 was determined to be 2α,3α,19α,23-tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester.
Compound 3 was isolated as a white powder. The HR-ESI-MS peak at m/z 781.4370 [M−H]− indicated the molecular formula of 3 to be C41H66O14, with one oxygen atom less than that of 2 (821.4290 [M+Na]+). The structure of the sugar chain was determined to be the same as that of 2 by comparison of their 1H- and 13C-NMR spectroscopic data (Table 1). Comparing the NMR spectra of 3 with those of 2 indicated that signals due to a hydroxymethyl group [δH 3.29, 3.50 (each, d, J=11.4 Hz)] at C-4 in 2 was replaced by a signal due to the α-methyl group [δH 1.20 (3H, s), δC 29.9] in 3, which was further confirmed by HMBC and NOESY experiments on 3. Thus, compound 3 was concluded to be 2α,3α,19α-trihydroxyolean-12-en-28-oic-acid 28-O-β-D-xylopyranosyl-(1→2)-β-D-glucopyranosyl ester.
Compound 4 was obtained as a white powder, with the molecular formula C36H56O11, as deduced from the [M+Na]+ peak at m/z 687.3729 by HR-ESI-MS. The 1H-NMR spectrum of 4 in CD3OD showed five methyl singlets (δH 0.78, 0.89, 1.02, 1.06, 1.67). Additional proton resonances observed included those ascribed to an olefinic proton at δH 5.53 (1H, t, J=3.6 Hz), two oxymethine protons at δH 3.61 (1H, d, J=2.4 Hz) and 3.90 (1H, m), two oxymethylene protons at δH 3.95 and 4.17 (1H each, d, J=11.4 Hz), and an anomeric proton signal at δH 5.40 (1H d, J=8.4 Hz) correlated in the HSQC spectrum with an anomeric carbons at δC 95.9 in the 13C-NMR spectrum (Table 1). The 13C-NMR spectrum of 4 displayed 36 carbon signals including five methyl (δC 16.9, 17.7, 17.9, 18.4, 22.6), four olefinic (δC 129.1, 129.2, 133.7, 138.3), of which δC 129.2 and 138.3 were typical of a double bond at C-12(13) of an ursane-type triterpene,11) and a carboxyl signal (δC 177.7) (Table 1). Comparing the NMR spectra of 4 with those of rubuside (4a)12) showed that 4 had two more OH groups. After several NMR experiments, including HMBC, heteronuclear multiple quantum coherence (HMQC), and NOESY, it was apparent that the signals due to α-methyl group at C-4 and methyl group at C-29 in 4 were replaced by signals due to two hydroxymethyl groups {[δH 3.39, 3.53 (each, d, J=10.2 Hz), and δC 71.3] and [δH 3.95, 4.17 (each, d, J=11.4 Hz), and δC 63.7]}. Acid hydrolysis of 4 with 2 M HCl afforded D-glucose, which was identified by GC analysis of its trimethylsilyl L-cysteine derivatives.10) In the HMBC spectrum of 4, a long-range correlation from Glc-H-1 (δH 5.40) to C-28 (δC 177.7) confirmed that the β-D-glucopyranosyl unit is attached to C-28. In the NOESY spectrum, NOE correlations were observed between the following proton pairs: H-2/H-24, H-2/H-3, H-2/H-25, indicating that hydroxyl group at C-2, C-3, and C-23 were all oriented to α position. Thus, compound 4 was characterized as 2α,3α,23,29-tetrahydroxyurs-12,19-dien-28-oic-acid 28-O-β-D-glucopyranosyl ester.
The structures of known compounds 5–7 were determined as luteolin (5),13) 5,4′-dihydroxyl-3,7,3′-trimethoxyflavone (6),14) and caffeic acid (7),15) respectively, by detailed spectroscopic analysis and comparison of their spectral data with reported values in the literatures.
Hepatoprotective activities against D-galactosamine-induced toxicity of the four new compounds (1–4) and three known compounds (5–7) were examined in WB-F344 cells. Compounds 1–3 were found to show hepatoprotective activity at 10−5 M in vitro. The hepatoprotective activity of 1 was more than that of bicyclol (10−5 M), a drug showing potent hepatoprotective activity.16) This is the first report of hepatoprotective activity from a Callicarpa L.
| Compound | x±s | Cell survival rate (% of normal) |
|---|---|---|
| Normal | 1.026±0.037 | 100 |
| Control | 0.325±0.076 | 28## |
| Bicyclolb) | 0.537±0.049 | 50* |
| 1 | 0.650±0.108 | 61* |
| 2 | 0.603±0.074 | 64* |
| 3 | 0.715±0.088 | 68**,△ |
| 4 | 0.324±0.067 | 28 |
| 5 | 0.276±0.105 | 31 |
| 6 | 0.159±0.066 | 19 |
| 7 | 0.182±0.051 | 22 |
a) Results are expressed as means±S.D. (n=3); ## p<0.001, significantly different from control by Student’s t-test; * p<0.05, ** p<0.01, significantly different from normal by Student’s t-test; △ p<0.05, significantly different from positive control by Student’s t-test. b) Positive control compound.
Optical rotations were measured on a Perkin-Elmer 341 polarimeter. The UV spectra were recorded in MeOH on a Perkin-Elmer Lambda 25 UV-Vis spectrophotometer. The 1H- (600 MHz), 13C- (150 MHz), and 2D-NMR spectra were recorded on a Bruker AVANCE III 600 instrument using tetramethylsilane (TMS) as an internal reference. ESI-MS data were collected on a MDS SCIEX API 2000 LC/MS/MS instrument. HR-ESI-MS data were obtained on an Acquity UPLC Xevo G2Q-Tof mass spectrometer. Preparative HPLC (high performance liquid chromatography) was conducted with an Agilent Technologies 1200 series instrument with a MWD detector using a YMC-pack ODS (Octadecylsilyl)-A column (5 µm, 250×20 mm) and a Shimadzu LC-6A instrument with a RID-10A refractive index detector using a XTerra prep MS C18 column (10 µm, 300×19 mm). Column chromatography was performed with silica gel 60 (100–200 mesh, Qingdao Marine Chemical Ltd., Qingdao, China) and Develosil ODS (75 µm, Nomura Chemical Co., Ltd., Japan). TLC (thin-layer chromatography) was carried out with glass precoated with silica gel GF254. Spots were visualized under UV light or by spraying with 10% sulfuric acid in EtOH followed by heating.
Plant MaterialThe leaves of Callicarpa nudiflora were collected from Wuzhishan, Hainan, China, in Aug. 2011 and identified by Prof. Guiping Yuan at Jiangxi Provincial Institute for Drug and Food Control, China. A voucher specimen (No. 20110817) has been deposited in the Herbarium of Jiangxi Provincial Institute for Drug and Food Control.
Extraction and IsolationThe powdered dried leaves of Callicarpa nudiflora (9.6 kg) were extracted three times with 80% EtOH under reflux (2 h each). The extracting solution was evaporated under reduced pressure to yield a dark brown residue (1.8 kg). The residue was suspended in water (15 L) and then successively partitioned with petroleum ether (3×15 L), EtOAc (3×15 L), and n-BuOH (3×15 L). After removing the solvent, the n-BuOH extract (545 g) was passed through a XAD-7 macroporous resin column eluted with H2O and H2O–EtOH (5 : 95, v/v), respectively. The H2O–EtOH (5 : 95, v/v) fraction (236 g) was separated by silica gel column chromatography (CC) using CHCl3–MeOH gradient mixtures (99 : 1–60 : 40, v/v) to give thirteen fractions (E1–E13). Fraction E5 (30.5 g) was subjected to silica gel CC and eluted with CHCl3–MeOH (90 : 10–70 : 30, v/v) to afford fifteen fractions (E5-1–E5-15). Fraction E5-11 (3.6 g) was separated by ODS CC (30–50%, MeOH–H2O) to give sixteen subfractions. Subfraction 15 (318 mg) was further separated by preparative HPLC (YMC-ODS-A 5 µm, 250×20 mm, detection at 210 nm) using CH3CN–H2O (19 : 81, v/v, 5 mL/min) containing 0.01% TFA as mobile phase to yield compounds 1 (9.0 mg) and 2 (8.5 mg). Fraction E5-9 (1.85 g) was chromatographed over reversed-phase silica gel, eluting with a gradient of increasing MeOH (25–45%) in H2O to give sixteen subfractions. Subfraction 16 (181 mg) was purified by preparative HPLC (XTerra prep MS C18 column 10 µm, 300×19 mm, detection at 210 nm) using CH3CN–H2O (19 : 81, v/v, 5 mL/min) containing 0.01% TFA as mobile phase to yield compounds 3 (7.6 mg) and 4 (9.0 mg). Subfraction 13 (240 mg) was subjected to preparative HPLC (YMC-ODS-A 5 µm, 250×20 mm, detection at 210 nm) using 20% CH3CN–H2O (7 mL/min) containing 0.05% TFA as mobile phase to yield compounds 5 (10.5 mg), 6 (132.7 mg), and 7 (12.0 mg).
2α,3α,19α,24-Tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-Glucopyranosyl Ester (1): White powder; [α]D20 −11.4 (c=0.12, MeOH); 1H-NMR (pyridine-d5, 600 MHz) and 13C-NMR (pyridine-d5, 150 MHz) see Table 1; positive ESI-MS m/z: 689.7 [M+Na]+; negative ESI-MS m/z: 665.6 [M−H]−; HR-ESI-MS m/z 665.3878 [M−H]− (Calcd for C36H57O11, 665.3842).
2α,3α,19α,23-Tetrahydroxyolean-12-en-28-oic-acid 28-O-β-D-Xylopyranosyl-(1→2)-β-D-glucopyranosyl Ester (2): White powder; [α]D20 −30.3 (c=0.03, MeOH); 1H-NMR (CD3OD, 600 MHz) and 13C-NMR (CD3OD, 150 MHz) see Table 1; positive ESI-MS m/z: 821.9 [M+Na]+; negative ESI-MS m/z: 834.7 [M+Cl]−; HR-ESI-MS m/z 821.4290 [M+Na]+ (Calcd for C41H66O15Na, 821.4294).
2α,3α,19α-Trihydroxyolean-12-en-28-oic-acid 28-O-β-D-Xylopyranosyl-(1→2)-β-D-glucopyranosyl Ester (3): White powder; [α]D20 −17.6 (c=0.06, MeOH); 1H-NMR (pyridine-d5, 600 MHz) and 13C-NMR (pyridine-d5, 150 MHz) see Table 1; positive ESI-MS m/z: 805.7 [M+Na]+; negative ESI-MS m/z: 781.5 [M−H]−; HR-ESI-MS m/z: 781.4370 [M−H]− (Calcd for C41H65O14, 781.4374).
2α,3α,23,29-Tetrahydroxyurs-12,19-dien-28-oic-acid 28-O-β-D-Glucopyranosyl Ester (4): White powder; [α]D20 −20.1 (c=0.04, MeOH); 1H-NMR (CD3OD, 600 MHz) and 13C-NMR (CD3OD, 150 MHz) see Table 1; positive ESI-MS m/z: 687.2 [M+Na]+; negative ESI-MS m/z: 663.6 [M−H]−; HR-ESI-MS m/z 687.3729 [M+Na]+ (Calcd for C36H56O11Na, 687.3720).
Determination of Absolute Configurations of the Sugar Moieties in 1–4The determination of the absolute configuration of the sugars in compounds 1–4 was conducted as described previously.7)
Protective Effect on Cytotoxicity Induced by D-Galactosamine in WB-F344 CellsThe hepatoprotective effects of compounds 1–7 were determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay17,18) in WB-F344 cells, with some modification. Each cell suspension of 1×104 cells in 200 µL of Dulbecco’s modified Eagle’s medium containing fetal calf serum (3%), penicillin (100 units/mL), and streptomycin (100 µg/mL) was planted in a 96-well microplate and precultured for 24 h at 37°C under a 5% CO2 atmosphere. Fresh medium (200 µL) containing bicyclol and test samples was added, and the cells were cultured for 1 h. Then, the cultured cells were exposed to 40 mM D-galactosamine for 24 h. Cytotoxic effects of test samples were measured simultaneously in the absence of D-galactosamine. The medium was changed into a fresh one containing 0.5 mg/mL MTT. After 3.5 h incubation, the medium was removed and 150 µL of dimethyl sulfoxide was added to dissolve formazan crystals. The optical density (OD) of the formazan solution was measured on a microplate reader at 492 nm. Inhibition (%) was obstained by the following formula:
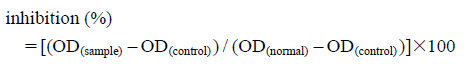 |
All values were expressed as ±S.D. The Student’s t-test for unpaired observations between normal or control and tested samples was carried out to identify statistical differences; p values less than 0.05 were considered as significantly different.
Financial support was provided by the National Natural Science Foundation of China (NSFC, Grant No. 81373955) and Fund for the Development of Youth in Inspection Institute of Research (No. 2013WA9). We thank Prof. Ai-Hong Liu at Center of Analysis and Testing Nanchang University for NMR measurements.