Notes
New Cassane-Type Diterpenoids from Caesalpinia bonduc
2014 Volume 62 Issue 7 Pages 729-733
Details
2014 Volume 62 Issue 7 Pages 729-733
Six new cassane diterpenoids, named caesalls H–M (1–6), were isolated from the seed kernels of Caesalpinia bonduc. Their structures were elucidated on the basis of spectroscopic analysis, mainly NMR and MS. The absolute configurations of compounds 1 and 3 were determined by a single-crystal X-ray study using a mirror CuKα radiation and circular dichroism (CD) spectra, respectively. None of the compounds were cytotoxic against HepG-2, MCF-7 and MG-63 cells.
Caesalpinia bonduc (LINN.) RINN. (Fabaceae), a rich source of cassane-type diterpenoids, is a stout prickly climber distributed throughout the tropical and subtropical regions. The structures of these diterpenoids are characterized by a molecular skeleton constructed of three fused cyclohexane rings and a furan ring or an α,β-butenolide moiety. Some cassane-type diterpenoids are known to possess antitumor, antimalarial, antibacterial, antihelmintic, and antineoplastic properties.1–5) Previous phytochemical investigations on plants of Caesalpinia have resulted in the isolation of several cassane and norcassane furanoditerpenes,6–9) and seven new compounds, caesalls A–G,10) were isolated in our previous research on C. bonduc. As a continuation of our study on this plant, the ethanol (95%) extract of the seed kernel was investigated. As a result, six new compounds (1–6) were obtained. Their structures were elucidated by extensive one dimensional (1D) and 2D NMR (heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond connectivity (HMBC), and rotating frame Overhauser enhancement spectroscopy (ROESY)) and mass (high resolution-electrospray ionization-mass spectrum (HR-ESI-MS)) spectroscopic data analysis. The absolute configuration of compound 1 was determined by a single-crystal X-ray diffraction experiment, and which of compound 3 was determined by circular dichroism (CD) spectra. None of the compounds were cytotoxic against HepG-2, MCF-7 and MG-63 cells. Herein, we reported the isolation and structure elucidation of compounds 1–6.

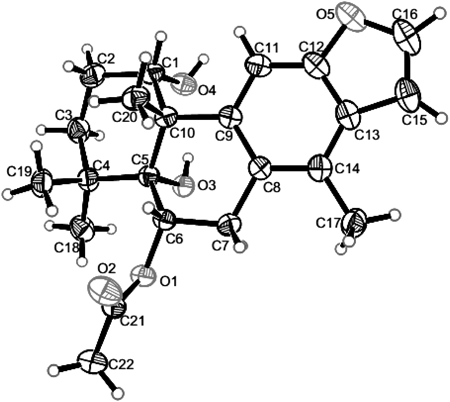
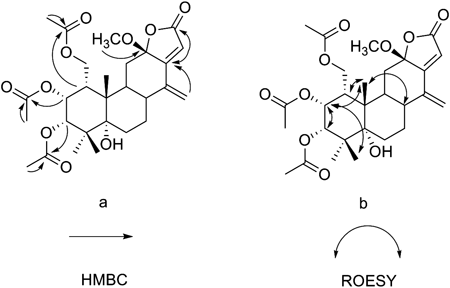
Compound 1 was recrystallized from CH2Cl2–MeOH to yield colorless crystals. A quasi-molecular peak [M+Na]+ at m/z 395.1830 in an HR-ESI-MS represents C22H28O5Na corresponded to a molecular formula of C22H28O5. The 1H-NMR spectrum showed signals corresponding to four tertiary methyls at δH 2.36 (H3-17), 1.28 (H3-18), 1.17 (H3-19) and δH 1.34 (H3-20), two oxygen-substituted methines at δH 4.56 (H-1), 5.71 (H-6), together with two protons of a 1,2-disubstituted furan ring (δH 7.55, d, J=2.0 and δH 6.74, d, J=2.0). In addition, the trisubstituted benzofuran moiety resonated the downfield signals at δH 7.55 (H-16), 6.74 (H-15), 7.40 (H-11), and one aromatic methyl group at δH 2.36 (H3-17). The 1D NMR data of compound 1 was closely related to these of 6-acetoxy-3-deacetoxycaesaldekarin e,2) except for the acetyl group at C-1 in 6-acetoxy-3-deacetoxycaesaldekarine that was replaced by hydroxyl group in 1. The locations of 6-OAc and 1-OH were deduced from HMBC correlations observed from a proton signal at δH 5.71 (H-6) to –OCOCH3 (δC 170.5) and C-5 (δC 78.2), and H3-20 (δH 1.34, s) to C-1 (δC 73.5), H-1 (δH 4.56, br s) to C-10 (δC 49.4). The relative stereochemistry of 1 was determined by ROESY spectral analysis. The methyl at δH 1.34 (H3-20) showed cross peaks with H-1 and H-6 indicated that the acetoxy group at C-6 and the hydroxyl group at C-1 were α-oriented. The absolute configuration of 1 was determined by X-ray diffraction study as shown in Fig. 2. Thus, the structure was confirmed and the absolute configuration of 1 was finally determined to be 1S,5R,6S,10S and named caesall H.
Compound 2 had a molecular formula C22H26O5, based on HR-ESI-MS ([M+H]+ at m/z 371.1850) which was 2 mass units less than that of 1. A comparison of the NMR data (Table 1) of compounds 1 and 2 showed that the hydroxyl group at C-1 in 1 was replaced by ketone carbonyls in 2. This difference was confirmed by the carbon signal at δC 211.0 and the HMBC correlation of H3-20 (δH 1.74, s) with C-1 (δC 211.0). In the ROESY spectrum, correlations from H-6 to H3-20 and H3-19 indicated that the acetoxy group at C-6 was α-oriented. Caesall I (2) is, therefore, the 1-keto analogue of 1 and a perspective view of the molecular structure is presented in Fig. 1.
| Position | 1 | δC | 2 | δC | 3 | δC | 4 | δC | 5 | δC | 6 | δC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δH (multi, J in Hz) | δH (multi, J in Hz) | δH (multi, J in Hz) | δH (multi, J in Hz) | δH (multi, J in Hz) | δH (multi, J in Hz) | |||||||
| 1 | 4.56, br s | 73.5 | 211.0 | 5.22, d (3.0) | 73.2 | 5.86, s | 74.5 | 5.86, s | 74.5 | 5.28, br s | 74.5 | |
| 2 | 1.93, m | 24.9 | 2.79, m | 36.5 | 5.50, t (3.0) | 65.9 | 5.47, dd (13.0, 3.0) | 68.0 | 5.46, dd (13.0, 3.0) | 68.0 | 5.32, ddd (13.0, 4.5, 3.0) | 67.3 |
| 2.16, m | ||||||||||||
| 3 | 1.21, m | 32.5 | 1.97, m | 39.0 | 5.15, d (3.0) | 77.3 | 2.19, dd (13.0, 5.0) | 36.1 | 2.16, dd (13.0, 5.0) | 36.1 | 1.39, dd (13.5, 4.5) | 35.4 |
| 2.16, m | 1.43, m | 1.43, m | 1.96, m | |||||||||
| 4 | 38.6 | 38.2 | 43.1 | 40.0 | 40.0 | 40.2 | ||||||
| 5 | 78.2 | 79.9 | 76.5 | 75.6 | 75.6 | 78.0 | ||||||
| 6 | 5.71, dd (9.0, 7.5) | 72.5 | 5.64, dd (9.5, 7.0) | 72.2 | 1.77, m | 26.2 | 1.99, m | 24.0 | 1.99, m | 23.9 | 1.60, dd (13.0, 5.5) | 32.0 |
| 1.68, m | 2.03, m | 2.03, m | 2.16, dd (13.0, 5.5) | |||||||||
| 7 | 2.86, dd (16.0, 9.0) | 31.9 | 2.80, m | 31.0 | 1.87, m | 23.0 | 2.65, dd (17.0, 7.0) | 23.7 | 2.65, dd (17.0, 7.0) | 23.6 | 5.20, td (11.0, 5.5) | 75.9 |
| 3.29, dd (16.0, 7.5) | 3.28, dd (16.0, 7.5) | 2.76, dd (17.0, 8.0) | 2.76, dd (17.0, 8.0) | |||||||||
| 8 | 127.3 | 124.5 | 2.09, m | 39.8 | 126.7 | 126.7 | 2.47, m | 38.9 | ||||
| 9 | 139.6 | 134.6 | 2.83, td (12.0, 2.5) | 37.9 | 142.4 | 142.4 | 2.61,td (12.0, 5.5) | 37.1 | ||||
| 10 | 49.4 | 60.0 | 45.5 | 48.4 | 48.4 | 45.1 | ||||||
| 11 | 7.40, s | 104.5 | 7.74, s | 111.3 | 1.43, m | 35.6 | 6.44, s | 102.9 | 6.43, s | 102.8 | 2.37, dd (16.0, 5.0) | 21.6 |
| 2.08, m | 2.50, m | |||||||||||
| 12 | 154.1 | 153.5 | 107.6 | 156.0 | 156.0 | 149.8 | ||||||
| 13 | 126.1 | 126.8 | 166.3 | 122.3 | 122.2 | 113.4 | ||||||
| 14 | 128.9 | 128.0 | 143.6 | 133.3 | 133.3 | 3.38, d (9.0) | 46.1 | |||||
| 15 | 6.74, d (2.0) | 105.2 | 6.71, d (2.0) | 105.0 | 5.90, s | 115.3 | 2.95, m | 37.4 | 2.95, m | 37.4 | 6.10, d (1.5) | 108.4 |
| 3.27, m | 3.27, m | |||||||||||
| 16 | 7.55, d (2.0) | 144.7 | 7.56, d (2.0) | 145.3 | 169.8 | 6.01, dd (7.0, 2.0) | 100.7 | 6.02, dd (7.0, 2.0) | 100.6 | 7.22, d (1.5) | 141.7 | |
| 17 | 2.36, s | 16.0 | 2.20, s | 16.2 | 5.21, d (2.0) | 113.6 | 2.12, s | 16.4 | 2.12, s | 16.4 | 174.6 | |
| 5.30, d (2.0) | ||||||||||||
| 18 | 1.28, s | 30.8 | 1.36, s | 29.0 | 1.12, s | 23.3 | 1.16, s | 28.0 | 1.16, s | 28.0 | 1.11, s | 28.2 |
| 19 | 1.17, s | 25.0 | 1.28, s | 26.6 | 1.21, s | 25.0 | 1.20, s | 25.7 | 1.20, s | 25.7 | 1.16, s | 25.9 |
| 20 | 1.34, s | 29.5 | 1.74, s | 28.8 | 1.17, s | 17.8 | 1.41, s | 30.2 | 1.41, s | 30.1 | 1.33, s | 17.7 |
| 1-OCOCH3 | 169.4 | 169.5 | 169.7 | 169.1 | ||||||||
| 1-OCOCH3 | 2.11, s | 20.9 | 1.99, s | 21.1 | 1.98, s | 21.1 | 2.12, s | 21.2 | ||||
| 2-OCOCH3 | 169.9 | 170.6 | 170.6 | 170.4 | ||||||||
| 2-OCOCH3 | 1.96, s | 20.7 | 2.02, s | 21.2 | 2.02, s | 21.2 | 1.97, s | 21.0 | ||||
| 3-OCOCH3 | 169.4 | |||||||||||
| 3-OCOCH3 | 2.10, s | 20.8 | ||||||||||
| 6-OCOCH3 | 170.5 | 170.3 | ||||||||||
| 6-OCOCH3 | 2.18, s | 22.0 | 2.09, s | 21.9 | ||||||||
| 7-OCOCH3 | 170.2 | |||||||||||
| 7-OCOCH3 | 1.98, s | 21.0 | ||||||||||
| 12-OCH3 | 3.08, s | 50.2 | ||||||||||
| 17-OCH3 | 3.71, s | 52.0 |
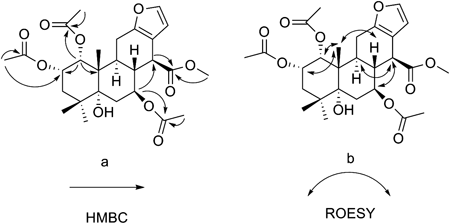
Compound 3, showed [M+Na]+ ion peak at m/z 543.2204 (calcd for C27H36O10Na 543.2201) and exhibited hydroxyl and carbonyl groups at 3455, 1749 cm−1 in the IR spectra. In the 1H- and 13C-NMR spectrum, the olefinic proton signal at δH 5.90 (H-15, s) and carbon signals at δC 107.6 (C-12), 115.3 (C-15), 166.3 (C-13) and 169.8 (C-16) suggested the presence of the α,β-butenolide moiety. The HMBC correlations (Fig. 3) of H-15 (δH 5.90, s) with C-13, C-16 confirmed the presence of such a moiety, combined with the three methyl signals at δH 1.12 (H3-18, s), 1.17(H3-20, s), 1.21(H3-19, s), which are typical signals for cassane-type diterpenes, that indicated 3 was a tetracyclic cassane diterpene possessing a fuesd butenolide unit. The obtained 1H- and 13C-NMR data were similar to caesalpin G,8) except for the absence of one hydroxyl group, the presence of two acetoxy groups and the difference of the relative configuration at C-12. Three acetoxy groups were attached at C-1, C-2, C-3 respectively, on the basis of the HMBC correlations from H-1 (δH 5.22, d, J=3.0) to –OCOCH3 (δC 169.4), from H-2 (δH 5.50, t, J=3.0) to –OCOCH3 (δC 169.9), from H-3 (δH 5.15, d, J=3.0) to –OCOCH3 (δC 169.4). The ROESY (Fig. 3) correlations of H3-20 with H-1, H-2 and H-8; H-2 with H-3 and H3-19 indicated that the acetoxy groups at C-1, C-2 and C-3 were α-oriented. Furthermore, the proton signal at δH 2.83 (H-9) showed no cross peaks with H3-20 and 12-OCH3 in ROESY spectrum indicated that H-9 was α-oriented and 12-OCH3 was β-oriented. The CD spectrum of 3 displayed a positive Cotton effect at 260 nm and a positive Cotton effect at 219 nm, confirming the (8R, 12S) absolute configuration for 3 based on the exciton chirality method and the γ-lactone rules.8,11–13) Thus, the structure of 3 was established as1α,2α,3α-triacetoxy-5α-hydroxy-12β-methoxycassa-14(17),13(15)-dien-16,12-olide and named caesall J.
Compounds 4/5 were isolated as a diasteromixture at C-16 position. The molecular formula of 4/5 was determined to be C24H32O7 by HR-ESI-MS (m/z 455.2043 [M+Na]+). The 1H-NMR spectrum (Table 1) of 4/5 indicated that it was an approximately 1 : 1 mixture of two stereoisomers, which we were unable to separate. Despite this, the low-field chemical shifts of H-11 (δH 6.44, s and 6.43, s), C-8 (δC 126.7, overlapping), C-9 (δC 142.4, overlapping), C-11 (δC 102.9 and 102.8), and C-14 (δC 133.3, overlapping) suggested that ring C in 4/5 is aromatic. The 1H-NMR spectrum displayed three methyl signals at δH 1.16 (H3-18, s), 1.20 (H3-19, s) and 1.41 (H3-20, s), two acetoxy methyl signals at δH 2.02, 1.98/1.97 and three oxymethines signals at δH 5.86 (H-1, s), 5.46/5.47 (H-2, d, J=13.0, 3.0) and 6.02/6.01 (H-16, d, J=7.0, 2.0). These data indicated that 4 was a tetracyclic cassane diterpene with two acetoxy substituents. Lack of fused furan ring signals and the presence of a oxymethine signal at δH 6.01/6.02, two protons signs at δH 2.95, 3.27 indicated the presence of a dihydrofuran ring. The location of H-16 was deduced from the HMBC correlations of H-16 (δH 6.01/6.02) with C-15 (δC 37.4), C-12 (δC 156.0), and C-13 (δC 122.2). The HMBC correlations (Fig. 4) of –OCOCH3 (δH 1.98, s and 1.99, s) with C-1 (δC 74.5, overlapping) and –OCOCH3 (δC 169.5, 169.7) and of –OCOCH3 (δH 2.02, overlapping) with C-2 (δC 68.0, overlapping) and –OCOCH3 (δC 170.6) indicated that the acetoxy groups were attached to C-1 and C-2. In the ROESY spectra, the methyl at δH 1.41 (H3-20) showed cross peaks with H-1 and H-2 indicated the acetyl substituents at C-1 and C-2 to be α-axially oriented. Therefore, the structures of 4/5 were determined and named caesall K/L.

Compound 6 had the molecular formula C27H36O10 determined by HR-ESI-MS. The 1H-NMR spectrum of 6 displayed signals corresponding to three tertiary methyls, three oxygen-substituted methines, three acetyl methyls, and a methoxy. Moreover, the 13C-NMR spectrum of 6 showed four olefinic carbons (δC 108.4, 113.4, 141.7, 149.8) and four oxygen-substituted carbons (δC 67.3, 74.5, 75.9, 78.0) together with four ester carbonyl carbons (δC 169.1, 170.2, 170.4, 174.6). The 1D NMR data (Table 1) of 6 were similar to those of caesalpinin MG,14) except for the presence of an acetoxy group at C-2 and the absence of the acetoxy group at C-6. The location of 2-OAc was confirmed by the HMBC relations (Fig. 5) of 1-H (δH 5.28, br s) with C-2 (δC 67.3) and of –COOCH3 (δH 1.97, s) with C-2 (δC 67.3) and –COOCH3 (δC 170.4). The relative configuration of 6 was assigned by the ROESY spectrum, the correlation of H3 −20 (δH 1.33, s) with H-1 (δH 5.28, br s) and H-2 (δH 5.32, ddd, J=13.0, 4.5, 3.0), of H-14 (δH 3.38, d, J=9.0) with H-7 (δH 5.20, td, J=11.0, 5.5) and H-9 (δH 2.61, td, J=12.0, 5.5) indicated that the acetoxyl substituents at C-1 and C-2 to be in α-oriented, and the acetoxyl substituents at C-7, C-14 to be β-oriented. Thus, the structure of caesall M was assigned as 6.
Compounds 1–6 were evaluated for their cytotoxicity against three human cancer cell lines (HepG-2, MCF-7 and MG-63) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. All compounds were inactive against all of the tested cell lines at a concentration of 100 µM.
Melting points (mp) were measured on an X-4 digital display micro-melting apparatus, uncorrected. Optical rotations were determined with a JASCO P-1020 polarimeter. UV spectra were performed on a Shimadzu UV-2450 spectrophotometer. IR spectra were recorded in KBr-disc on a Bruker Tensor 27 spectrometer. 1D and 2D NMR spectra were acquired on a Bruker AV-500 NMR instrument at 500 MHz (1H) and 125 MHz (13C) in CDCl3. HR-ESI-MS was carried out on an Agilent UPLC-Q-TOF (6520B). Column chromatography (CC) was done using silica gel (Qingdao marine Chemical Co., Ltd., China), ODS (40–63 µm, FuJi, Japan), or Sephadex LH-20 (Pharmacia, Sweden). Preparative HPLC was carried out using a Shimadzu LC-6A instrument with a SPD-10A detector using a shim-pack RP-C18 column (20×200 mm). Analytical HPLC was measured on an Agilent 1200 Series instrument with a DAD detector using a shim-pack VP–ODS column (250×4.6 mm).
Plant MaterialThe seeds of Caesalpinia bonduc (LINN.) ROXB. (Fabaceae) were purchased from Chengdu City, Sichuan province of China in March 2012, and were authenticated by Professor Min-Jian Qin, Department of Medicinal Plants, China Pharmaceutical University. A voucher specimen (No. CC201203) was deposited in the Department of Natural Medicinal Chemistry, China Pharmaceutical University.
Extraction and IsolationThe powdered air-dried seed kernels of C. bonduc (2.8 kg) were extracted with 95% EtOH (3×4 h). The EtOH extract was concentrated under reduced pressure. The crude extract (790 g) was suspended in water and successively partitioned with petroleum ether, CH2Cl2. The petroleum ether fraction, the CH2Cl2 fraction yielded 360 g, 142 g after removal of the solvent, respectively. Fractionation of the CH2Cl2 extract was performed on a silica gel column using a gradient of petroleum ether–acetone (50 : 1, 25 : 1, 10 : 1, 3 : 1) to yield four fractions 1–4 by TLC analysis. Fraction C2 (7 g) was run on an octadecyl silica (ODS) column using a step gradient of MeOH–H2O (60 : 40 to 100 : 0), to afford four subfractions (C2.1–2.4). Fraction C2.4 was purified by preparative HPLC using the mobile phase MeOH–H2O (70 : 30) to yield 1 (20 mg). Fraction C2.2 was chromatographed over a Sephadex LH-20 column, eluted with MeOH to give 4/5 (9 mg). Fraction C2.1 was chromatographed over an ODS column with a continuous gradient of MeOH–H2O (50 : 50 to 100 : 0) to afford 6 (1.5 mg). Fraction C2.3 was eluted with MeOH–H2O (70 : 30) on an ODS column and further purified by preparative HPLC using the mobile phase MeOH–H2O (70 : 30) to yield 2 (3 mg).
Caesall H (1): colorless crystals; mp 204–206°C; [α]D25 +79.6° (c 0.05, MeOH); UV (MeOH) λmax (log ε) 211 (5.65), 250 (5.28), 280 (4.54), 291 (4.55) nm; IR (KBr) νmax 3454, 1729, 1639, 1402, 1244 cm−1; 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 395.1830 [M+Na]+ (Calcd for C22H28NaO5: 395.1829).
Caesall I (2): White powder; [α]D25 +114.6° (c 0.07, MeOH); UV (MeOH) λmax (log ε) 210 (5.35), 250(5.02), 280 (4.40), 290(4.37) nm; IR (KBr) νmax 3444, 2320, 1638, 1400, 1241, 1045 cm−1; 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 371.1850 [M+H]+ (Calcd for C22H27O5: 371.1853).
Caesall J (3): White powder; [α]D25 −23.3° (c 0.08, MeOH); UV (MeOH) λmax (log ε) 202 (3.03), 216 (sh) (2.70) nm; CD (MeOH) 215 (Δε+9.0), 233 (Δε+4.7), 260 (Δε+12.4) nm; IR (KBr) νmax 3455, 2355, 1749, 1637, 1402, 1256 cm−1; 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 543.2204 [M+Na]+ (Calcd for C27H36NaO10: 543.2201).
Caesall K/L (4/5, 1 : 1 diastereomixture): White powder; [α]D25 −4.9° (c 0.20, MeOH); UV (MeOH) λmax (log ε) 205 (4.21), 282 (2.30) nm; IR (KBr) νmax 3453, 1743, 1640, 1399, 1385, 1250, 1036 cm−1; 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 455.2043 [M+Na]+ (Calcd for C24H32NaO7: 455.2040).
Caesall M (6): White powder; [α]D25 −17.0° (c 0.10, MeOH); UV (MeOH) λmax (log ε) 215 (3.93) nm; IR (KBr) νmax 3578, 3467, 2955, 1742, 1367, 1241 cm−1; 1H- and 13C-NMR data, see Table 1; HR-ESI-MS m/z 543.2198 [M+Na]+ (Calcd for C27H36NaO10: 543.2201).
X-Ray Crystallographic AnalysisColorless crystals of 1 were obtained from CH2Cl2–MeOH. Crystal data were obtained on a Bruker Smart-1000 CCD with a graphite monochromator with CuKα radiation (λ=1.54184 Å) at 290(2) K. The structure was solved by direct methods using the SHELXS-9715) and expanded using difference Fourier techniques, refined with the SHELXL-97.16) Crystal data of 1: C22H28O5 (M=372.44); monoclinic crystal (0.37×0.36×0.3 mm3); space group P21; unit cell dimensions a=10.7688(2) Å, b=7.7077(10) Å, c=11.7281(3) Å, β=99.691(2)°, V=959.57(3) Å3; Z=2; Dcalcd=1.289 mg/m3; µ=0.733 mm−1; 7749 reflections measured (7.64≤2Θ≤139.32); 3385 unique (Rint=0.0200) which were used in all calculations; the final refinement gave R1=0.0332 (>2sigma(I)) and wR2=0.0950 (all data); flack parameter=0.04(16). Crystallographic data for compound 1 have been deposited in the Cambridge Crystallographic Data Centre (deposition number: CCDC 981573). Copies of the data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK. [fax: (+44)1223–336–033 or e-mail: deposit@ccdc.cam.ac.uk].
BioassaysThe cytotoxicity of compounds 1–6 was assessed via the MTT method using the HepG-2, MG-63 and MCF-7 cancer cell lines. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and cultured at a density of 5000 cell/mL in a 96-well microtiter plate. Five different concentrations of each compound in dimethyl sulfoxide (DMSO) were subsequently added to the wells. Each concentration was tested in triplicate. After incubation under 5% CO2 at 37°C for 48 h, 20 µL of MTT (4 mg/mL) was added to each well, and the cells were incubated for another 4 h. Then, the liquid in each well was removed, and DMSO (150 µL) was added. The absorbance (OD values) at 570 nm with a 630 nm reference was measured on a Universal Microplate Reader.
This research work was financially supported by the National Natural Sciences Foundation of China (81202899), the National High Technology Research and Development Program of China (863 Program) (2013AA093001), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT-IRT1193).