Experimental
General MethodsAll reagents were purchased from Sigma-Aldrich Chemicals Co., Tokyo Kasei Co., Wako Pure Chemical Industries, Ltd., and Kanto Chemical Co., Inc. Silica gel column chromatography was purchased from Kanto Chemical Co., Inc. 1H-NMR spectra were recorded at 400 MHz on a Brucker Avance 400 spectrometer or 500 MHz on a Brucker Avance 500 spectrometer. 13C-NMR spectra were recorded on at 125 MHz on a Brucker Avance 500 spectrometer. Chemical shifts are reported as parts per million (ppm) relative to chloroform (7.26 ppm for 1H-NMR and 77.16 ppm for 13C-NMR) or dimethylsulfoxide (DMSO) (2.50 ppm for 1H-NMR and 39.52 ppm for 13C-NMR). Data are reported as follows; chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; sex, sextet; sep, septet; br, broad; m, multiplet) coupling constants (Hz), integration. Melting points were taken on a Yanagimoto micro melting point apparatus. Mass spectral data were obtained with a JEOL AX-505 spectrometer. Elemental analyses were carried out on a Yanaco MT-6 CHN CORDER.
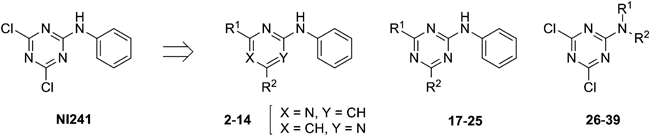
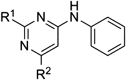
2,6-Dichloro-4-phenylaminopyrimidine (2) and 4,6-Dichloro-2-phenylaminopyrimidine (3)Aniline (1.0 mL, 10.90 mmol) was added dropwise to 2,4,6-trichloropyrimidine (2.00 g, 10.90 mmol) in tetrahydrofuran (30 mL). The mixture was stirred at room temperature for 3 h, then Et3N (2.27 mL, 16.35 mmol) and aniline (0.50 mL, 5.45 mmol) were further added in one portion. The mixture was stirred at room temperature for 18 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 10 : 1 to 4 : 1) gave 2 (70%) and 3 (25%). 2: colorless crystals (dichloromethane–hexane); mp 143.2–144.6°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.45 (td, 2H, J=7.0, 0.6 Hz), 7.30 (tt, 1H, J=7.5, 1.0 Hz), 7.28 (dd, 2H, J=7.4, 1.2 Hz), 7.15 (br s, 1H), 6.57 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ: 163.3, 161.2, 160.1, 136.2, 129.9, 126.8, 123.8, 100.7. Anal. Calcd for C10H7Cl2N3: C, 50.03; H, 2.94; N, 17.50. Found: C, 50.00; H, 2.96; N, 17.39. 3: colorless needles (hexane); mp 115.1–116.9°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.57 (dd, 2H, J=8.7, 1.1 Hz), 7.36 (td, 2H, J=7.1, 1.0 Hz), 7.23 (br s, 1 H), 7.11 (tt, 1H, J=7.4, 1.1 Hz), 6.79 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ: 161.8, 158.9, 137.7, 129.1, 124.0, 119.7, 111.2. Anal. Calcd for C10H7Cl2N3: C, 50.03; H, 2.94; N, 17.50. Found: C, 50.16; H, 3.08; N, 17.46.
4-Chloro-2-methoxy-6-phenylaminopyrimidine (4)Sodium methoxide (58.5 mg, 1.08 mmol) was added to 2 (200 mg, 0.83 mmol) in methanol (4.2 mL) at 0°C. The mixture was stirred at room temperature for 4 h. The mixture was stirred at 50°C for 2 h. The contents were poured into saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 4 (32%) as colorless needles (dichloromethane–hexane); mp 174.2–176.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.40 (td, 2H, J=7.4, 0.9 Hz), 7.32 (dd, 2H, J=8.6, 1.7 Hz), 7.22 (tt, 1H, J=7.3, 1.2 Hz), 6.97 (br s, 1H), 6.35 (s, 1H), 3.96 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 165.2, 163.6, 161.2, 137.4, 129.5, 125.6, 123.2, 96.8, 55.0. Anal. Calcd for C11H10ClN3O: C, 56.06; H, 4.28; N, 17.83. Found: C, 56.07; H, 4.34; N, 17.88.
2,4-Diisopropoxy-6-phenylaminopyrimidine (5)Sodium hydride (60% dispersion in mineral oil, 66.6 mg, 1.67 mmol) in 2-propanol (6 mL) was added to 2 (100 mg, 0.42 mmol) under Ar. The mixture was stirred at room temperature for 1 h and at 100°C for 24 h, then sodium hydride (60% dispersion in mineral oil, 16.66 mg) suspended in 2-propanol (3 mL) was further added in one portion. The resulting mixture was stirred at 100°C for 24 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel flash chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 5 (67%) as colorless needles (hexane); mp 86.1–88.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.57 (dd, 2H, J=8.7, 1.1 Hz), 7.25 (dd, 2H, J=7.1, 0.8 Hz), 7.12 (tt, 1H, J=7.4, 1.2 Hz) 6.54 (br s, 1H), 5.32 (sep, 1H, J=6.2 Hz), 5.21 (sep, 1H, J=6.3 Hz), 1.38 (d, 6H, J=6.4 Hz), 1.30 (d, 6H, J=6.0 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 171.7, 164.5, 163.6, 138.8, 129.3, 124.3, 122.4, 80.9, 69.5, 68.7, 22.1, 22.0. Anal. Calcd for C15H18N3O2: C, 66.88; H, 7.37; N, 14.62. Found: C, 66.82; H, 7.19; N, 14.72.
4-Chloro-6-methoxy-2-phenylaminopyrimidine (6)Sodium methoxide (58.50 mg, 1.08 mmol) was added to 3 (200 mg, 0.83 mmol) in methanol (4 mL) at 0°C. The mixture was stirred at room temperature for 23 h, then poured into saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, and evaporated. Recrystallization of the residue gave 6 (quant.) as colorless plates (hexane); mp 105.0–105.9°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.59 (dd, 2H, J=8.7, 1.1 Hz), 7.34 (td, 2H, J=7.1, 1.0 Hz), 7.22 (br s, 1 H), 7.07 (tt, 1H, J=7.4, 1.1 Hz), 6.22 (s, 1H), 3.97 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.2, 160.6, 159.0, 138.8, 128.9, 123.1, 119.4, 97.9, 54.3. Anal. Calcd for C11H10ClN3O: C, 56.06; H, 4.28; N, 17.83. Found: C, 56.26; H, 4.31; N, 17.82.
4-Chloro-6-ethoxy-2-phenylaminopyrimidine (7)Sodium hydride (60% dispersion in mineral oil) (33.6 mg, 0.84 mmol) in ethanol (6 mL) was added to 3 (70 mg, 0.29 mmol) at 0°C under Ar. The mixture was stirred at room temperature for 5 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 7 (quant.) as pale-yellow needles (hexane); mp 109.1–111.2°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.57 (dd, 2H, J=8.8, 1.2 Hz), 7.33 (td, 2H, J=7.2, 2.0 Hz), 7.06 (tt, 1H, J=7.6, 0.8 Hz), 7.08–7.04 (br s, 1H), 6.20 (s, 1H), 4.41 (q, 2H, J=7.2 Hz), 1.40 (t, 3H, J=7.2 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 170.9, 159.6, 158.5, 138.5, 129.0, 123.2, 119.5, 98.2, 63.4, 14.3. Anal. Calcd for C12H12ClN3O: C, 57.72; H, 4.84; N, 16.83. Found: C, 57.65; H, 4.80; N, 16.93.
4,6-Diisopropoxy-2-phenylaminopyrimidine (8)Sodium hydride (60% dispersion in mineral oil, 45.5 mg, 1.14 mmol) in 2-propanol (3 mL) was added to 3 (136.4 mg, 0.57 mmol) at 0°C under Ar. The mixture was stirred at room temperature for 1 h and at 100°C for 24 h, then sodium hydride (60% dispersion in mineral oil, 68.17 mg, 1.70 mmol) suspended in 2-propanol (3 mL) was further added in one portion. The resulting mixture was stirred at 100°C for 24 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel flash chromatography (eluent: hexane–ethyl acetate 20 : 1) gave 8 (56%) as a colorless oil; 1H-NMR (CDCl3, 400 MHz) δ: 7.59 (dd, 2H, J=8.8, 1.2 Hz), 7.30 (td, 2H, J=8.0, 1.2 Hz), 7.00 (tt, 1H, J=7.4, 1.2 Hz), 6.83 (br s, 1H), 5.48 (s, 1H), 5.23 (sep, 2H, J=7.4 Hz), 1.35 (d, 12 H J=6.4 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 171.1, 158.8, 139.8, 128.7, 122.0, 118.8, 82.4, 69.0, 22.0.
4-Chloro-2-(4-methoxybenzyl)amino-6-phenylaminopyrimidine (9)4-Methoxybenzylamine (68.6 mg, 0.50 mmol) and Et3N (63.3 mg, 0.63 mmol) were added to 2 (100 mg, 0.42 mmol) in THF (4 mL) at 0°C. The mixture was stirred at room temperature for 18 h and at 50°C for 4 h, then 4-methoxybenzylamine (28.6 mg, 0.21 mmol) and Et3N (21.1 mg, 0.21 mmol) were further added in one portion. The resulting mixture was stirred at 50°C for 3 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and then concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 9 (88%). 9: colorless crystals (hexane); mp 143.8–146.3°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.35 (td, 2H, J=7.0, 2.3 Hz), 7.30 (dd, 2H, J=8.6, 1.5 Hz), 7.26 (dd, 2H, J=6.1, 2.0 Hz), 7.15 (tt, 1H, J=7.0, 1.5 Hz), 6.87 (dd, 2H, J=6.6, 2.1 Hz), 6.57 (br s, 1 H), 6.04 (s, 1H), 5.27 (s, 1H), 4.53 (d, 2H, J=5.8 Hz), 3.80 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 162.4, 161.8, 160.4, 158.8, 138.0, 130.9, 129.3, 128.7, 124.7, 122.4, 114.0 93.1, 55.3, 44.9. Anal. Calcd for C18H17ClN4O: C, 63.44; H, 5.03; N, 16.44. Found: C, 63.28; H, 5.03; N, 16.45.
2-Amino-4-chloro-6-phenylaminopyrimidine (10)Trifluoroacetic acid (1.0 mL) was added to a solution of 9 (34.6 mg, 0.10 mmol). The mixture was stirred at room temperature for 25 h, then poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 2 : 1) gave 10 (quant.) as colorless powder (ethyl acetate–hexane); mp 209.6–210.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.38 (td, 2H, J=8.0, 0.8 Hz), 7.30 (dd, 2H, J=8.8, 1.6 Hz), 7.19 (tt, 1H, J=7.4, 1.2 Hz), 6.59 (br s, 1 H), 6.11 (s, 1 H), 4.90 (br s, 2H); 13C-NMR (CDCl3, 125 MHz) δ: 162.8, 162.4, 160.7, 137.6, 129.5, 125.2, 122.9, 93.8. Anal. Calcd for C10H9ClN4: C, 54.43; H, 4.11; N, 25.39. Found: C, 54.38; H, 4.22; N, 25.15.
4-Chloro-6-(4-methoxybenzylamino)-2-phenylaminopyrimidine (11)4-Methoxybenzylamine (57.1 mg, 0.42 mmol) and Et3N (31.6 mg, 0.31 mmol) were added to 3 (50 mg, 0.20 mmol) in THF (2 mL). The mixture was stirred at room temperature for 1 h and at 50°C for 14 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 2 : 1) gave 11 (81%) as a colorless oil; 1H-NMR (CDCl3, 400 MHz) δ: 7.53 (dd, 2H, J=8.4, 0.8 Hz), 7.27 (td, 2H, J=7.2, 2.0 Hz), 7.24 (dd, 2H, J=6.8, 2.0 Hz), 7.00 (tt, 1H, J=8.0, 1.2 Hz), 7.02–6.98 (br s, 1H), 6.89 (dd, 2H, J=6.8, 2.0 Hz), 5.88 (s, 1H), 5.18 (br s, 1H), 4.46 (br s, 2H), 3.80 (s, 3 H); 13C-NMR (CDCl3, 125 MHz) δ: 163.7, 159.2, 139.2, 128.8, 112.5, 119.3, 114.2, 55.3, 53.4, 45.2, 45.1.
4-Amino-6-chloro-2-phenylaminopyrimidine (12)Trifluoroacetic acid (2.0 mL) was added to a solution of 11 (92.0 mg, 0.42 mmol). The mixture was stirred at room temperature for 17 h, then poured into saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 1 : 1) gave 2–13 (quant.) as colorless plates (dichloromethane–hexane); mp 139.2–141.0°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.55 (dd, 2H, J=8.8, 1.2 Hz), 7.31 (td, 2H, J=7.6, 2.0 Hz), 7.04 (tt, 1H, J=7.2, 1.2 Hz), 6.91 (br s, 1H), 5.97 (s, 1H), 4.81 (br s, 2H); 13C-NMR (CDCl3, 125 MHz) δ: 164.3, 160.0, 159.6, 139.0, 128.8, 122.8, 119.6, 95.0. Anal. Calcd for C10H9ClN4: C, 54.43; H, 4.11; N, 25.39. Found: C, 54.29; H, 4.26; N, 25.52.
4-Chloro-2-methyl-6-phenylaminopyrimidine (14)Aniline (560 µL, 6.13 mmol) was added dropwise to 4,6-dichloro-2-methylpyrimidine (1.0 g, 6.13 mmol) in ethanol (30 mL) at 0°C. The mixture was stirred at room temperature for 19 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel flash chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 14 (53%) as colorless needles (hexane); mp 109.1–111.2°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.42 (td, 2H, J=7.6, 0.8 Hz), 7.24 (dd, 2H, J=8.8, 1.2 Hz), 7.24 (tt, 1H, J=7.6, 0.8 Hz), 6.87 (br s, 1H), 6.54 (s, 1H), 2.54 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 167.43, 161.1, 160.9, 136.5, 129.9, 129.6, 129.2, 126.3, 123.4, 99.9, 25.2. Anal. Calcd for C11H10ClN3: C, 60.14; H, 4.59; N, 19.13. Found: C, 60.36; H, 4.79; N, 19.15.
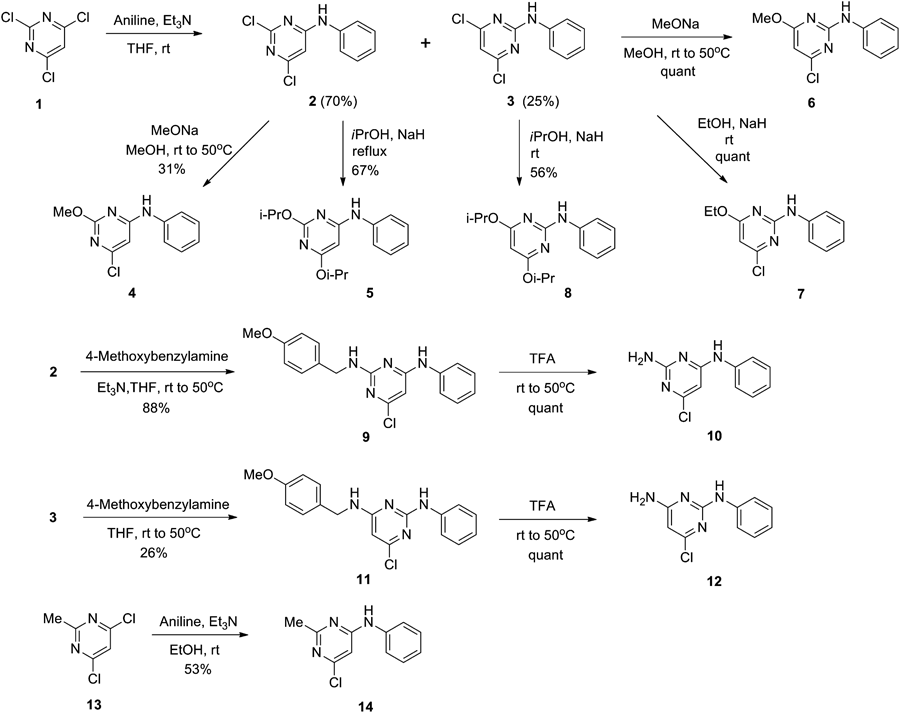
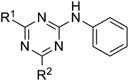
2,4-Dichloro-6-phenylamino-1,3,5-triazine (16) and 2-Chloro-4,6-bis(phenylamino)-1,3,5-triazine (17)Aniline (0.98 µL, 10.85 mmol) was added dropwise to cyanuric chloride (2.0 g, 10.85 mmol) in acetone (60 mL) at 0°C. Et3N (1.5 mL, 10.85 mmol) was added in one portion. The mixture was stirred at room temperature for 7 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 8 : 1 to 6 : 1) gave 16 (53%) and 17 (10%). 16: colorless crystals (ethyl acetate–hexane); mp 134.1–140.6°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.54 (dd, 2H, J=8.8, 1.2 Hz), 7.49 (br s, 1H), 7.42 (td, 2H, J=7.2, 5.6 Hz), 7.23 (tt, 1H, J=7.6, 1.2 Hz). 13C-NMR (CDCl3, 125 MHz) δ: 171.4, 170.2, 164.1, 135.7, 129.3, 125.9, 121.2. Anal. Calcd for C9H6Cl2N4: C, 44.84; H, 3.51; N, 23.24. Found: C, 45.13; H, 2.69; N, 23.43. 17: colorless crystals (ethyl acetate–hexane); mp 201.8–204.0°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.54 (d, 4H, J=8.0 Hz), 7.36 (t, 4H, J=7.6 Hz), 7.29 (br s, 2H), 7.16 (t, 2H, J=7.4 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 168.6, 164.0, 137.0, 129.1, 121.4, 121.4. Anal. Calcd for C15H12ClN5: C, 60.51; H, 4.06; N, 23.52. Found: C, 60.75; H, 4.18; N, 23.65.
2-Chloro-4-methoxy-6-phenylamino-1,3,5-triazine (18)Sodium methoxide (22.1 mg, 0.41 mmol) was added to 16 (100 mg, 0.41 mmol) in methanol (2.0 mL) at 0°C. The mixture was stirred at room temperature for 18 h. Sodium methoxide (11.1 mg, 0.21 mmol) was further added in one portion and stirring was continued at room temperature for 3 h, then the mixture was poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 7 : 1) gave 18 (78%) as colorless needles (ethyl acetate–hexane); mp 174.2–176.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.56 (dd, 2H, J=8.8, 1.2 Hz), 7.38 (td, 2H, J=7.6, 2.0 Hz), 7.41–7.36 (br s, 1H), 7.17 (tt, 1H, J=7.6, 0.8 Hz), 4.04 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.6, 170.7, 165.2, 136.7, 129.2, 124.9, 120.8, 55.8. Anal. Calcd for C10H9ClN4O: C, 50.75; H, 3.83; N, 23.67. Found: C, 50.74; H, 3.94; N, 23.88.
2,4-Di-n-butoxy-6-phenylamino-1,3,5-triazine (19)Sodium hydride (60% dispersion in mineral oil, 18.0 mg, 0.45 mmol) in 1-butanol (2 mL) was added to 16 (101 mg, 0.42 mmol) at 0°C under Ar. The mixture was stirred at room temperature for 1 h and at 40°C for 1 h, then sodium hydride (60% dispersion in mineral oil, 33.4 mg, 0.84 mmol) suspended in 1-butanol (2 mL) was further added in one portion. The resulting mixture was stirred at 40°C for 4 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 10 : 1) gave 19 (86%) as colorless plates (hexane); mp 58.4–59.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.58 (dd, 2H, J=8.8, 1.2 Hz), 7.34 (td, 2H, J=7.2, 1.2 Hz), 7.10 (tt, 1H, J=7.4, 1.6 Hz), 7.12–7.08 (br s, 1H), 4.38 (t, 4H), 1.78 (quint, 4H, J=7.2 Hz), 1.48 (sex, 4H, J=7.4 Hz), 0.96 (t, 4H, J=7.4 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 172.0, 166.32, 137.9, 128.9, 123.8, 120.4, 67.7, 30.7, 19.1, 13.8. Anal. Calcd for C17H24N4O2: C, 64.53; H, 7.65; N, 17.71. Found: C, 64.45; H, 7.37; N, 17.80.
2-Chloro-4-phenylamino-6-phenoxy-1,3,5-triazine (20)Sodium hydride (60% dispersion in mineral oil, 20.1 mg, 0.49 mmol) suspended in N,N-dimethylformamide (DMF) (1 mL) was added to phenol (67.3 mg, 0.72 mmol) and 16 (101 mg, 0.42 mmol) in DMF at 0°C under Ar. The mixture was stirred at 0°C for 2 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 2 : 1) gave 20 (73%) as colorless crystals (hexane); mp 137.1–138.8°C; 1H-NMR (DMSO-d6, 373 K, 400 MHz) δ: 10.36 (br s, 1H) 7.50–7.45 (m, 4H), 7.32 (tt, 1H, J=7.4, 1.2 Hz), 7.28 (dd, 2H, J=9.2, 1.6 Hz), 7.23 (t, 1H, J=7.6 Hz), 7.07 (t, 2H, J=7.4 Hz); Anal. Calcd for C15H11ClN4O: C, 60.31; H, 3.71; N, 18.76. Found: C, 60.53; H, 3.85; N, 18.87.
2-Chloro-4-methylamino-6-phenylamino-1,3,5-triazine (21)Methylammonium chloride (70 mg, 0.49 mmol) and Et3N (70 µL, 0.50 mmol) were added to 16 (101 mg, 0.42 mmol) in 2-propanol (2 mL) at 0°C. The mixture was stirred at room temperature for 4 h, and then methylammonium chloride (4 mg, 0.06 mmol) was further added in one portion. The resulting mixture was stirred at room temperature for 1 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 2 : 1) gave 21 (53%) as a colorless powder (ethyl acetate); mp 243.9–245.3°C; 1H-NMR (DMSO-d6, 373 K, 400 MHz) δ: 9.57 (br s, 1H), 7.69 (d, 2H, J=8.0 Hz), 7.60 (br s, 1H), 7.29 (t, 2H, J=8.0 Hz), 7.02 (t, 1H, J=7.2 Hz), 2.84 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 168.9, 168.1, 166.2, 164.1, 163.6, 139.4, 129.1, 123.3, 120.5, 17.9, 27.8. Anal. Calcd for C10H10ClN5: C, 50.96; H, 4.28; N, 29.72. Found: C, 51.17; H, 4.45; N, 29.74.
2-Chloro-4-dimethylamino-6-phenylamino-1,3,5-triazine (22)Dimethylammonium chloride (33.0 mg, 0.40 mmol) and Et3N (57 µL, 0.41 mmol) were added to 16 (100.2 mg, 0.41 mmol) in 2-propanol (2 mL) at 0°C. The mixture was stirred at room temperature for 4 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 10 : 1) gave 22 (85%) as colorless needles (ethyl acetate–hexane); mp 162.2–163.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.57 (dd, 2H, J=8.8, 1.2 Hz), 7.34 (td, 2H, J=7.2, 2.0 Hz), 7.09 (tt, 1H, J=7.4, 1.2 Hz), 7.00 (br s, 1H), 3.20 (s, 6H); 13C-NMR (CDCl3, 125 MHz) δ: 169.0, 165.2, 163.4, 138.0, 128.9, 123.7, 120.1, 36.8, 36.7. Anal. Calcd for C11H12ClN5: C, 52.91; H, 4.82; N, 28.05. Found: C, 52.80; H, 4.99; N, 28.10.
2-Chloro-4-phenylamino-6-(pyrrolidin-1-yl)-1,3,5-triazine (23)Pyrrolidine (45 µL, 0.54 mmol) was added to a solution of 16 (101 mg, 0.42 mmol) in dichloromethane (2 mL) at 0°C. The mixture was stirred at room temperature for 15 min, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 7 : 1) gave 23 (34%) as colorless needles (ethyl acetate–hexane); mp 191.0–192.0°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.40 (td, 2H, J=7.4, 0.9 Hz), 7.33 (dd, 2H, J=7.2, 2.0 Hz), 7.08 (tt, 1H, J=7.6, 1.2 Hz), 7.00 (br s, 1H), 3.63 (t, 4H, J=6.8 Hz), 1.99 (tt, 4H, J=3.4, 3.2 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 168.7, 163.2, 1629, 138.2, 128.9, 123.5, 119.8, 46.8, 46.6, 25.2, 25.0. Anal. Calcd for C11H12ClN5: C, 56.63; H, 5.12; N, 25.40. Found: C, 56.62; H, 5.17; N, 25.43.
2-Methylamino-4-methoxy-6-phenylamino-1,3,5-triazine (24)Methylammonium chloride (36.1 mg, 0.42 mmol) and Et3N (70 µL, 0.50 mmol) were added to 18 (99.9 mg, 0.42 mmol) in methanol (2 mL) at 0°C. The mixture was stirred at room temperature for 2 h and at 40°C for 4 h, then methylammonium chloride (34.5 mg, 0.51 mmol) and Et3N (70 µL, 0.50 mmol) were further added. The resulting mixture was refluxed for 1 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 24 (88%) as colorless crystals (ethyl acetate–hexane); mp 163.1–164.5°C; 1H-NMR (DMSO-d6, 373 K, 400 MHz) δ: 9.06 (br s, 1H), 7.73 (d, 2H, J=8.0 Hz), 7.25 (t, 2H, J=7.8 Hz), 7.01 (br s, 1H), 6.95 (t, 1H, J=7.4 Hz), 3.83 (s, 3H), 2.84 (d, 3 H J=4.8 Hz); Anal. Calcd for C11H13N5O: C, 57.13; H, 5.67; N, 30.28. Found: C, 57.31; H, 5.65; N, 30.28.
2-Dimethylamino-4-methoxy-6-phenylamino-1,3,5-triazine (25)Dimethylammonium chloride (69.0 mg, 0.85 mmol) and Et3N (120 µL, 0.80 mmol) were added to a solution of 18 (101 mg, 0.43 mmol) in methanol (2 mL) at 0°C. The mixture was stirred at room temperature for 2 h and at 50°C for 2 h, then methylammonium chloride (38.2 mg, 0.47 mmol) and Et3N (60.0 µL 0.43 mmol) were added in one portion. The resulting mixture was refluxed for 1 h, then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. Purification of the residue by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) gave 25 (78%) as colorless needles (ethyl acetate–hexane); mp 163.1–164.5°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.61 (dd, 2H, J=8.8, 1.2 Hz), 7.32 (td, 2H, J=7.2, 2.0 Hz), 7.04 (tt, 1H, J=7.2, 1.2 Hz), 6.85 (br s, 1H), 3.94 (s, 3H), 3.19 (s, 6H). 13C-NMR (CDCl3, 125 MHz) δ: 170.9, 166.6, 165.0, 139.0, 128.8, 122.8, 119.8, 54.0, 36.6, 36.3. Anal. Calcd for C12H15N5O: C, 58.76; H, 6.16; N, 28.55. Found: C, 58.90; H, 6.04; N, 28.66.
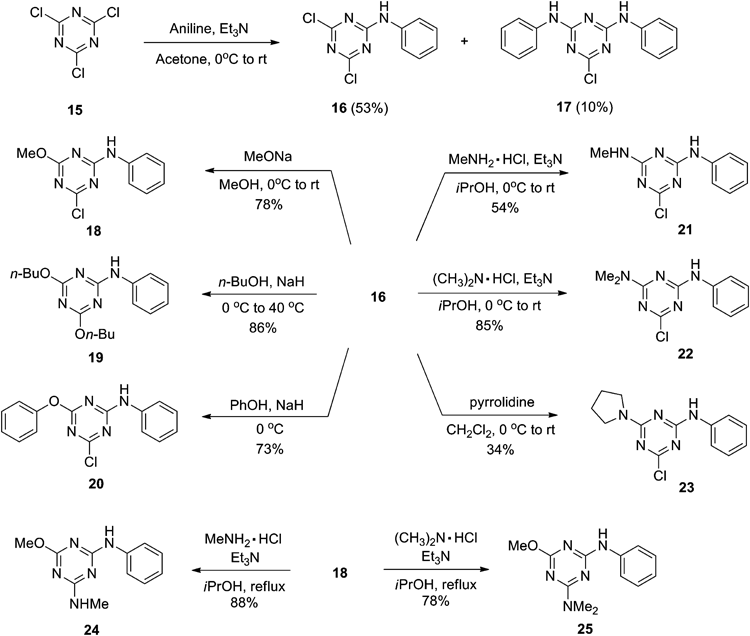
2,4-Dichloro-6-methyl(phenyl)amino-1,3,5-triazine (26)N-Methylaniline (58 µL, 0.54 mmol) and Et3N (75 µL, 0.54 mmol) were added dropwise to a solution of cyanuric chloride (101 mg, 0.55 mmol) in acetone (2 mL) at 0°C under Ar. The mixture was stirred at room temperature for 2 h, and then poured into water. The precipitate was collected by filtration, washed with water, dried, and recrystallized to gave 26 (91%) as colorless prisms (CH2Cl2–hexane); mp 134.0–132.7°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.46 (td, 2H, J=7.8, 2.0 Hz), 7.36 (tt, 1H, J=7.4, 2.0 Hz), 7.24 (dd, 2H, J=7.2, 1.2 Hz), 3.55 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 170.3, 170.0, 165.1, 142.0, 129.5, 127.8, 126.1, 39.2. Anal. Calcd for C10H8Cl2N4: C, 47.12; H, 3.33; N, 21.85. Found: C, 47.08; H, 3.16; N, 21.96.
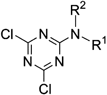
General Procedure for the Synthesis of Compounds 27–29Anisidine (1 eq) was added dropwise to a solution of cyanuric chloride (1 eq) in acetone (2 mL) at 0°C. The mixture was stirred at room temperature for 2 h. After the completion of the reaction, the mixture was evaporated. The crude product was purified by silica gel chromatography (eluent: hexane–ethyl acetate 5 : 1) and then recrystallized.
2,4-Dichloro-6-(2-methoxyphenyl)amino-1,3,5-triazine (27): Yield: 86%; pale-yellow plates (ethyl acetate–hexane); mp 179.4–181.2°C; 1H-NMR (CDCl3, 400 MHz) δ: 8.31 (dd, 1H, J=8.0, 1.6 Hz), 8.20 (br s, 1H), 7.15 (td, 1H, J=7.8, 1.2 Hz), 7.04 (td, 1H, J=7.8, 1.2 Hz), 6.94 (dd, 1H, J=8.0, 1.2 Hz), 3.91 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.4, 170.1, 148.9, 125.7, 125.5, 121.2, 120.9, 110.5, 56.0. Anal. Calcd for C10H8Cl2N4O: C, 44.30; H, 2.97; N, 20.67. Found: C, 44.24; H, 3.09; N, 20.71.
2,4-Dichloro-6-(3-methoxyphenyl)amino-1,3,5-triazine (28): Yield: 72%; colorless needles (dichloromethane–hexane); mp 116.7–117.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.50 (br s, 1H), 7.30 (t, 1H, J=8.2 Hz), 7.26 (t, 1H, J=2.4 Hz), 7.05 (ddd, 1H, J=8.0, 2.0, 0.8 Hz), 6.77 (ddd, 1H, J=8.4, 2.4. 0.8 Hz), 3.84 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.5, 170.4, 164.1, 160.4, 137.0, 130.2, 113.4, 111.3, 107.4, 55.6. Anal. Calcd for C10H8Cl2N4O: C, 44.30; H, 2.97; N, 20.67. Found: C, 44.68; H, 3.25; N, 20.32.
2,4-Dichloro-6-(4-methoxyphenyl)amino-1,3,5-triazine (29): Yield: 67%; pale-yellow plates (ethyl acetate–hexane); mp 170.0–173.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.43–7.40 (br s, 1H), 7.41 (dd, 2H, J=6.8, 2.4 Hz), 6.93 (dd, 2H, J=6.8, 2.4 Hz), 3.82 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.5, 170.2, 164.4, 128.5, 123.6, 114.6, 55.7. Anal. Calcd for C10H8Cl2N4O: C, 44.30; H, 2.97; N, 20.67. Found: C, 47.43; H, 3.26; N, 20.66.
General Procedure for the Synthesis of Compounds 30–35 and 38Amine (1 eq) was added dropwise to cyanuric chloride (1.2 eq) in acetone at 0°C. The mixture was stirred at room temperature for 1 h then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude product was purified by silica gel chromatography (eluent: hexane–ethyl acetate) and then recrystallized.
2,4-Dichloro-6-benzylamino-1,3,5-triazine (30): Yield: 34%; colorless needles (dichloromethane–hexane); mp 120.1–121.3°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.40 (m, 5H), 6.15 (br s, 1H), 4.68 (d, 2H, J=6.0 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 171.3, 170.3, 166.0, 136.3, 129.2, 128.4, 127.9, 45.6. Anal. Calcd for C10H8Cl2N4: C, 47.08; H, 3.16; N, 21.96. Found: C, 47.05; H, 3.22; N, 21.99.
2,4-Dichloro-6-benzyl(methyl)amino-1,3,5-triazine (31): Yield: 48%; colorless needles (hexane); mp 99.4–101.6°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.38–7.30 (m, 3H), 7.26–7.23 (m, 2H), 4.87 (s, 2H), 3.15 (s, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 170.5, 170.2, 165.3, 135.5, 129.1, 128.3, 128.0, 52.8, 35.0. Anal. Calcd for C11H10Cl2N4: C, 49.09; H, 3.75; N, 20.82. Found: C, 49.19; H, 3.64; N, 20.92.
2,4-Dichloro-6-diethylamino-1,3,5-triazine (32): Yield: 54%; colorless crystals (hexane); mp 78.0–78.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 3.63 (q, 4H, J=7.2 Hz), 1.22 (t, 6H, J=7.2 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 169.0, 163.0, 41.6, 11.7. Anal. Calcd for C7H10Cl2N4: C, 38.03; H, 4.56; N, 25.34. Found: C, 38.30; H, 4.56; N, 25.52.
2,4-Dichloro-6-t-butylamino-1,3,5-triazine (33): Yield: 65%; colorless plates (hexane); mp 129.4–132.0°C; 1H-NMR (CDCl3, 400 MHz) δ: 5.78 (br s, 1H), 1.46 (s, 9H).
2,4-Dichloro-6-n-hexylamino-1,3,5-triazine (34): Yield: 56%; colorless needles (hexane); mp 54.8–55.7°C; 1H-NMR (CDCl3, 400 MHz) δ: 3.47 (q, 2H, J=6.8 Hz), 1.60 (quint, 2H, J=7.2 Hz), 1.40–1.26 (m, 6H), 0.90 (t, 3H, J=6.8 Hz).
2,4-Dichloro-6-cyclohexylamino-1,3,5-triazine (35): Yield: 40%; colorless oil; 1H-NMR (CDCl3, 400 MHz) δ: 5.76 (br s, 1H), 3.93–3.88 (m, 1H), 2.01–1.96 (m, 2H), 1.77–1.70 (m, 2H), 1.67–1.61 (m, 1H), 1.47–1.36 (m, 2H), 1.29–1.20 (m, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.1, 170.0, 165.1, 50.3, 32.6, 25.4, 24.5. Anal. Calcd for C9H12Cl2N4: C, 47.08; H, 3.16; N, 21.96. Found: C, 47.05; H, 3.22; N, 21.99.
2,4-Dichloro-6-(piperidin-1-yl)-1,3,5-triazine (38): Yield: 40%; colorless plates (hexane); mp 88.9–89.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 3.81 (t, 4H, J=5.6 Hz), 1.75–1.69 (m, 2H), 1.67–1.61 (m, 4H); 13C-NMR (CDCl3, 125 MHz) δ: 170.3, 163.7, 45.5, 25.8, 24.4. Anal. Calcd for C8H10Cl2N4: C, 41.22; H, 4.32; N, 23.98. Found: C, 41.19; H, 4.68; N, 24.04.
General Procedure for the Synthesis of Compounds 36, 37 and 39Amine (1 eq) in tetrahydrofuran was added dropwise to cyanuric chloride (1.2 eq) and K2CO3 (1 eq) in tetrahydrofuran at 0°C. The mixture was stirred at room temperature for 3–4 h then poured into water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated. The crude product was purified by silica gel chromatography (eluent: hexane–ethyl acetate) and then recrystallized.
2,4-Dichloro-6-(furfurylamino)-1,3,5-triazine (36): Yield: 92%; colorless needles (hexane–ethyl acetate); mp 104.7–105.4°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.38 (dd, 1H, J=1.6, 0.4 Hz), 6.35 (dd, 1H, J=3.2, 2.0 Hz), 6.32 (dd, 1H, J=3.2, 0.8 Hz), 6.20 (br s, 1H), 4.67 (d, 2H, J=6.0 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 171.2, 170.3, 165.8, 149.3, 143.0, 110.1, 108.7, 38.5. Anal. Calcd for C8H6Cl2N4O: C, 39.21; H, 2.47; N, 22.86. Found: C, 39.28; H, 2.49; N, 22.98.
2,4-Dichloro-6-(1-naphthylamino)-1,3,5-triazine (37): Yield: 93%; colorless needles (hexane–ethyl acetate); mp 156.7–157.8°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.92–7.80 (m, 5H), 7.60–7.52 (m, 3H); 13C-NMR (CDCl3, 125 MHz) δ: 171.8, 170.7, 165.9, 134.4, 130.5, 129.0, 128.2, 128.0, 127.3, 126.7, 125.7, 122.9, 120.9. Anal. Calcd for C13H8Cl2N4: C, 53.63; H, 2.77; N, 19.24. Found: C, 53.58; H, 2.98; N, 19.26.
2,4-Dichloro-6-(1,2,3,4-tetrahydroquinolin-1-yl)-1,3,5-triazine (39): Yield: 93%; orange needles (hexane–ethyl acetate); mp 159.2–160.6°C; 1H-NMR (CDCl3, 400 MHz) δ: 7.72 (d, 1H, J=8.0 Hz), 7.27–7.16 (m, 3H),4.07 (t, 2H, J=6.4 Hz), 2.83 (t, 2H, J=6.8 Hz), 2.05 (quint, 2H, J=6.5 Hz); 13C-NMR (CDCl3, 125 MHz) δ: 170.5, 170.4, 164.1, 136.7, 132.4, 129.0, 126.2, 126.0, 125.1, 45.7, 26.8, 23.6. Anal. Calcd for C12H10Cl2N4: C, 51.27; H, 5.59; N, 19.93. Found: C, 51.38; H, 3.60; N, 20.08.
Electrophoretic Mobility Shift Assay (EMSA)Alexa680-labeled NF-κB probe was prepared by annealing with Alexa680-labeled single-stranded oligonucleotide and unlabeled complementary single-stranded oligonucleotide: 5′-Alexa680-AGTTGAGGGGACTTTCCCAGGC-3′ (sense) and 5′-GCCTGGGAAAGTCCCCTCAACT-3′ (antisense). The underlines show the sequence of the κB site. His-tagged NF-κB p50 recombinants were produced in E. coli and purified with HisTrap HP (GE Healthcare). Reaction mixtures containing binding buffer (15 mM Tris–HCl (pH 7.5), 75 mM NaCl, 1.5 mM ethylenediamine tetraacetic acid (EDTA), 1.5 mM dithiothreitol, 7.52% glycerol, and 0.3% Nonidet P-40), 0.5 µg of poly(dI-dC)·(dI-dC) and 62.5 ng of His-tagged p50 recombinants were left to stand on ice for 10 min, and then the candidate inhibitors were added and the mixtures were incubated at room temperature. After 30 min, 20 nM Alexa680 labeled NF-κB probe was added and incubation was continued at room temperature for 30 min. The samples in a volume of 10 µL were loaded on native 5% polyacrylamide gel prepared in 0.5×TBE and electrophoresed at 120 CV for 90 min. The gels were scanned and analyzed with an Odyssey Infrared Imaging System (LI-COR).